Background
Denys-Drash syndrome (DDS) is a rare disorder consisting of the triad of congenital nephropathy, Wilms tumor, and intersex disorders resulting from mutations in the Wilms tumor suppressor (WT1) gene. Nephropathy is a constant feature; in the incomplete forms of the syndrome, the nephropathy is present with either Wilms tumor or intersex disorders, but the vast majority of patients with Denys-Drash syndrome are destined to develop Wilms tumor in any residual renal tissue. See the image below.
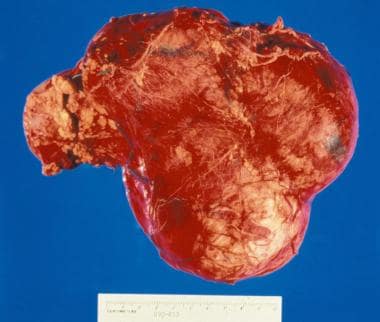 Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side.
Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side.
See Wilms Tumor: A Pediatric Oncology Success Story, a Critical Images slideshow, to help identify the clinical features, staging evaluation, prognostic factors, and therapeutic options for this disease.
The characteristic nephropathy in Denys-Drash syndrome is termed diffuse mesangial sclerosis. [1] This condition clinically manifests as an early onset nephrotic syndrome and progresses to renal failure during the first 3 years of life. Among the intersex disorders, pure gonadal dysgenesis with male pseudohermaphroditism is the classic presentation, although a wide variety of abnormalities in gonadal differentiation can be encountered.
Pathophysiology
Denys-Drash syndrome is the result of mutations in the WT1 gene on chromosome band 11p13. [2, 3, 4, 5] The WT1 gene contains 10 exons that produce 4 different messenger RNAs (mRNAs) as a result of 2 alternative splicing sites in exons 5 and 9 that, in turn, encode 4 different isoforms of the WT1 protein. Splicing at the second alternative site (exon 9) is thought to have a great biological importance and results in the inclusion or exclusion of 3 amino acids, lysine, threonine, and serine (KTS), yielding the KTS-positive isoform when the amino acids are included and KTS-negative isoform when excluded. The precise ratio of the KTS-positive/negative isoforms seems to be crucial for the normal function of the WT1 gene.
The WT1 protein is a transcription factor predominantly expressed in the embryonic kidneys and gonads. Exons 1-6 of the WT1 gene encode the regulatory domain, which regulates expression of target genes, and exons 7-10 encode the 4 zinc fingers of the DNA-binding region of the WT1 protein. The WT1 protein mediates the mesenchymal-epithelial transition and differentiation during morphogenesis of the kidney and gonad by repressing genes that encode cell proliferation factors and by activating genes that encode markers of epithelial cell differentiation.
Point mutations in the WT1 gene result in loss of its regulatory function, with the consequent abnormalities in glomerular formation and gonadal differentiation seen in Denys-Drash syndrome. Mutations that disrupt the second alternative splicing site of the WT1 gene alter the normal ratio of KTS-positive/negative isoforms from 2:1 to 1:2 and result in abnormalities in glomerular formation and gonadal differentiation seen in Frasier syndrome. In striking contrast, complete deletions of band 11p13 result in the Wilms tumor, aniridia, genitourinary malformations, and mental retardation (WAGR) syndrome, which is characterized by structural urinary tract abnormalities without nephropathy.
Epidemiology
Frequency
International
The frequency of Denys-Drash syndrome is unknown. Worldwide, more than 200 cases of Denys-Drash syndrome have been reported since 1967, when Denys et al originally described a child with nephropathy, ambiguous genitalia, and Wilms tumor. [6, 7, 8] New cases are being reported worldwide. [9]
Mortality/Morbidity
Mortality and morbidity are high because of the natural history of the nephropathy and the high risk of malignancies. Note the following:
-
Nephropathy: Patients with Denys-Drash syndrome develop early-onset nephrotic syndrome, have a high prevalence of severe hypertension, and experience rapid progression to end-stage renal disease (ESRD).
-
Malignancy: The vast majority of patients with Denys-Drash syndrome are destined to develop Wilms tumor in the native kidneys and are at significant risk for development of gonadoblastoma in the dysgenetic gonads.
Race-, sex-, and age-related demographics
Denys-Drash syndrome has no race predilection.
Although both sexes can be affected, the presence of intersex disorders makes the estimation of the male-to-female ratio misleading because individuals with Denys-Drash syndrome who are assigned the female gender may be genotypic males (XY gonadal dysgenesis with female phenotype). Ascertainment is also biased toward children with ambiguous genitalia (males), whereas diagnosis in females may be delayed or not established.
Nephropathy: Nephrotic syndrome usually manifests in infants aged 2 weeks to 18 months. Progression to ESRD occurs within weeks to 2 years from the time of diagnosis or before the age of 3 years.
Wilms tumor: Median age at discovery is 12.5 months in cases associated with Denys-Drash syndrome, as opposed to 36 months in patients with isolated Wilms tumor without Denys-Drash syndrome. The earliest tumor onset was in patients with truncation mutations (12 mo, 66 patients) compared with missense mutations (18 mo, 30 patients).
Intersex disorders: These conditions usually manifest at birth.
Prognosis
All patients develop nephropathy and progress to ESRD within 2 years from the diagnosis or before age 3 years.
Virtually all patients with Denys-Drash syndrome who have their native kidneys develop Wilms tumor. Patients with unilateral Wilms tumor are at risk for contralateral tumor. Staging and histologic criteria are prognostic in patients with Wilms tumor.
Patient Education
Provide genetic counseling.
Explain role of prophylactic surgery to prevent Wilms tumor and gonadoblastoma.
Explain renal replacement therapy options, including renal transplantation.
-
Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side.