Background
Glycogen-storage disease (GSD) type III (GSD III) is an autosomal recessive inborn error of metabolism caused by loss of function mutations of the glycogen debranching enzyme (Amylo-1,6-glucosidase [AGL]) gene, which is located at chromosome band 1p21.2. [1] GSD III is characterized by the storage of structurally abnormal glycogen, termed limit dextrin, in both skeletal and cardiac muscle and/or liver, with great variability in resultant organ dysfunction. [2, 3, 4]
In 1928, Snappes and van Creveld provided the first description of 2 patients with GSD III (see Online Mendelian Inheritance in Man [OMIM]). Both patients had hepatomegaly and reduced ability to mobilize hepatic glycogen stores.
In 1953, Forbes provided an extensive clinical description of a third patient with GSD III and suggested that the glycogen in both liver and muscle tissues had an abnormal structure. [5] Illingworth and Cori isolated the glycogen from the tissues of this patient and showed that it had extremely short outer chains. [6] This structure had previously been termed a limit dextrin by Cori and Cori, specifically to identify a glycogen molecule that had been extensively hydrolyzed by phosphorylase (ie, the enzyme that cleaves the alpha1,4-glycosidic bonds that form the linear backbone of glycogen) but that contained all of the alpha1,6-glycosidic bonds that formed the branch points of the original glycogen molecule. Cori and Cori predicted that the patient's condition was caused by a debranching enzyme deficiency. [7] See the image below.
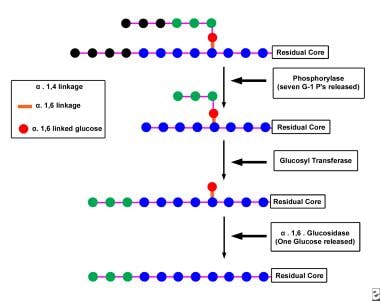 Schematic illustration of the degradation of glycogen by the concerted action of the enzymes phosphorylase and debranching enzyme. First, phosphorylase removes glucose moieties (linked to their neighbors via alpha1,4 glucosidic bonds and depicted as the 7 black circles) from the unbranched outer portions of the glycogen molecule until only 4 glucosyl units (depicted as the 3 green circles and the 1 red circle) remain before an alpha1,6 branch point. The transferase component of debranching enzyme then transfers the 3 (green) glucose residues from the short branch to the end of an adjacent branch of the glycogen molecule. The glucosidase component of debranching enzyme then removes the glucose moiety (depicted as the red circle) remaining at the alpha1,6 branch point. In the process, the branch point formed by the alpha1,6 glucosidic bond is removed, hence the name debrancher.Unlike phosphorylase, which removes glucose moieties from glycogen in the form of glucose-1-phosphate, debrancher releases 1 free glucose moiety from each branch point. After the cleavage of the branch site, phosphorylase attacks unbranched portions of the glycogen molecule until the enzyme is stymied by the appearance of another branch point, at which point debranching enzyme once again is called into play. Eventually, large portions of the glycogen molecule are degraded to free glucose by the action of the amylo-alpha1,6-glucosidase activity of debranching enzyme and to glucose-1-phosphate by the action of phosphorylase.
Schematic illustration of the degradation of glycogen by the concerted action of the enzymes phosphorylase and debranching enzyme. First, phosphorylase removes glucose moieties (linked to their neighbors via alpha1,4 glucosidic bonds and depicted as the 7 black circles) from the unbranched outer portions of the glycogen molecule until only 4 glucosyl units (depicted as the 3 green circles and the 1 red circle) remain before an alpha1,6 branch point. The transferase component of debranching enzyme then transfers the 3 (green) glucose residues from the short branch to the end of an adjacent branch of the glycogen molecule. The glucosidase component of debranching enzyme then removes the glucose moiety (depicted as the red circle) remaining at the alpha1,6 branch point. In the process, the branch point formed by the alpha1,6 glucosidic bond is removed, hence the name debrancher.Unlike phosphorylase, which removes glucose moieties from glycogen in the form of glucose-1-phosphate, debrancher releases 1 free glucose moiety from each branch point. After the cleavage of the branch site, phosphorylase attacks unbranched portions of the glycogen molecule until the enzyme is stymied by the appearance of another branch point, at which point debranching enzyme once again is called into play. Eventually, large portions of the glycogen molecule are degraded to free glucose by the action of the amylo-alpha1,6-glucosidase activity of debranching enzyme and to glucose-1-phosphate by the action of phosphorylase.
Only 4 years later, Illingworth, Cori, and Cori demonstrated the enzyme deficiency in GSD III in 1956. Thirty-six years after his initial 1928 report, van Creveld, with the aid of Huijing, demonstrated deficient debranching activity in his original patients. [8] The clinical status of both patients had significantly improved since their conditions were originally described in 1928. Although Snappes and van Creveld's patients with GSD III were the first individuals in whom a defect in glycogen metabolism was reported, Cori and Cori demonstrated in 1952 that the absence of glucose-6-phosphatase activity was the enzyme defect in GSD I (von Gierke disease). Indeed, GSD I was the first inborn error of metabolism in which the precise enzyme defect was identified.
Since 1952, the various GSDs have been categorized numerically by the chronologic order in which the enzymatic defects were identified. The sole exception to this general rule is GSD type 0, which is not a true GSD because the quantity of liver glycogen in this condition is less than the amount in healthy individuals. Moreover, the hepatic glycogen in this condition has an entirely normal chemical structure.
In recognition of their pioneering work on the structure, synthesis, and metabolism of glycogen, the husband and wife team of Carl F. Cori and Gerty T. Cori were corecipients of the 1947 Nobel Prize in Physiology or Medicine. This award came 5 years before they had defined the nature of the enzymatic defect in GSD I.
A great deal of misinformation surrounds the incidence and clinical characteristics of GSD III, including the following:
-
First, GSD I is widely believed to be the most common of the GSDs, and GSD III is considered relatively rare. In fact, 2 large studies have demonstrated GSD I, GSD III, and GSD VI incidences to be approximately equal. Collectively, these 3 types account for approximately 80% of GSD cases. GSD II, which is actually a lysosomal storage disease, accounts for an additional 15% of all GSD cases. Moreover, because GSD III can have a mild clinical presentation, its incidence may be underestimated.
-
Second, a common belief is that the hypoglycemia experienced by patients with GSD I is much more severe than the hypoglycemia experienced by patients with GSD III. Although this usually is true, many patients with GSD III have hypoglycemia as severe or even more severe than the hypoglycemia experienced in GSD I.
-
Third, the hepatic signs and symptoms in GSD III are widely believed to always improve with advancing age and to possibly even disappear after puberty. In fact, a significant number of patients with GSD III develop overt cirrhosis and even liver failure after puberty; moreover, in several patients, the cirrhosis has progressed to aggressive hepatocellular carcinoma. [9]
-
Fourth, approximately 85% of patients with GSD III have significant involvement of both the liver and the skeletal muscles, a form of the disease referred to as GSD IIIa, which is a fact that is often overlooked. Muscular involvement usually is minimal during childhood but often becomes the predominant feature by young adulthood. [2] Progressive muscle weakness and distal muscle wasting frequently become disabling as the patients enter the third or fourth decade of life, although this condition has been reported to begin in childhood in many Japanese patients.
-
Fifth, muscular involvement can also involve the heart. Cardiomegaly is not rare, and a few patients have developed a dilated hypertrophic cardiomyopathy. [2] The disorder is confined to the liver in only 15% of patients with GSD III.
Pathophysiology
Understanding the clinical abnormalities of GSD III requires familiarity with the structure and function of both glycogen and glycogen debranching enzyme.
Role and availability of glycogen
The polysaccharide glycogen is a readily mobilized storage form of glucose. Because glucose is the primary energy source for most mammalian cells, the survival advantages of having a readily available storage form of this carbohydrate are obvious. Although glycogen serves this essential function in virtually every organ, the liver and the skeletal muscles are the major sites of glycogen storage. Smaller concentrations of glycogen are found in almost every tissue, even in the brain.
Although the concentration of glycogen per gram of tissue is much higher in the liver than in muscle, the total amount of glycogen stored in muscle is much larger than the amount stored in the liver because of the relative masses of the 2 organs. The importance of this energy resource can be appreciated by noting that the free glucose content of the body fluids of a child who weighs 10 kg is approximately 5 g, whereas tissue glycogen content, even after fasting 10 hours, is approximately 25 g.
Glycogen structure
Glycogen is a highly branched polymer of glucose in which most of the glucose residues are linked to each other by alpha1,4-glycosidic bonds to form a linear backbone (see the image below).
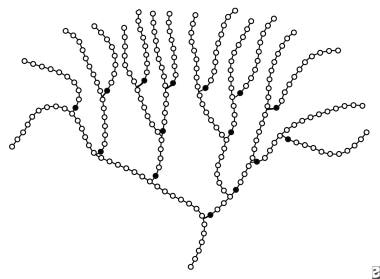 Schematic representation of a portion of a molecule of glycogen. Open circles represent the glucose moieties connected to each other via alpha1,4 linkages. Solid circles represent the glucose moieties connected to their neighbors via alpha1,6 linkages. Thus, each solid circle represents a branch point in the molecule.
Schematic representation of a portion of a molecule of glycogen. Open circles represent the glucose moieties connected to each other via alpha1,4 linkages. Solid circles represent the glucose moieties connected to their neighbors via alpha1,6 linkages. Thus, each solid circle represents a branch point in the molecule.
Interspersed along the linear backbone, at intervals of 4-10 glucose residues, are branches created by alpha1,6-glycosidic bonds. As a result of the extensive branching, the glycogen molecule has a frondlike and highly branched configuration with an open helical tertiary structure. [6] The helix, in turn, is organized into spherical particles with a molecular weight of 10-15 million (60,000 glucose residues per particle), and the spherical particles, in turn, are organized into large granules. The granules range in size from 10-40 nm and are located in the cellular cytosol.
Function of glycogen
Although glycogen is most abundant in liver and muscle, it has 2 quite different primary functions in these tissues. In muscle, glycogen is employed as a fuel source (ie, a source for the production of ATP) during brief periods of high energy consumption. In contrast, glycogen's major role in the liver is as a key player in the complex process of glucose homeostasis.
During times of energy abundance (eg, after a meal), the liver takes up glucose and nutrients that it can convert into glucose (primarily amino acids, galactose, fructose, lactate, pyruvate, and glycerol, but not fatty acids) from the bloodstream and converts these nutrients to glycogen. Conversely, when blood glucose levels fall, the liver catabolizes glycogen to glucose via a series of exquisitely regulated hydrolytic reactions referred to as glycogenolysis. Glucose is then available for delivery to tissues that cannot synthesize the carbohydrate in significant quantities (eg, brain, muscle, erythrocyte).
Not surprisingly, the predominant features of GSDs that primarily involve muscle are muscle cramps, exercise intolerance, easy fatigability, progressive weakness, and myopathy; in some cases, cardiomyopathy is a feature. In contrast, the predominant features of GSDs that primarily involve the liver are hepatomegaly, hepatic dysfunction, and hypoglycemia.
Debranching enzyme
Debranching enzyme (usually called debrancher) is a large protein composed of 1,532 amino acids organized as a single polypeptide with a molecular mass of approximately 170,000 daltons. This enzyme is unusual in that it is among the few proteins with 2 independently functioning catalytic activities located at separate sites on a single polypeptide chain. The 2 catalytic activities of debranching enzyme are a transferase, oligo-1,4-1,4-glucanotransferase (EC 2.4.1.25), and a glucosidase, amylo-alpha1,6-glucosidase (EC 3.2.1.33). Complete degradation of glycogen requires the concerted action of the enzymes phosphorylase and of both debranching enzyme components.
Phosphorylase first removes glucose moieties (see the image below), which are linked to their neighbors via alpha1,4-glucosidic bonds from the unbranched outer portions of the glycogen molecule until only 4 glucosyl units remain before an alpha1,6 branch point.
Schematic illustration of the degradation of glycogen by the concerted action of the enzymes phosphorylase and debranching enzyme. First, phosphorylase removes glucose moieties (linked to their neighbors via alpha1,4 glucosidic bonds and depicted as the 7 black circles) from the unbranched outer portions of the glycogen molecule until only 4 glucosyl units (depicted as the 3 green circles and the 1 red circle) remain before an alpha1,6 branch point. The transferase component of debranching enzyme then transfers the 3 (green) glucose residues from the short branch to the end of an adjacent branch of the glycogen molecule. The glucosidase component of debranching enzyme then removes the glucose moiety (depicted as the red circle) remaining at the alpha1,6 branch point. In the process, the branch point formed by the alpha1,6 glucosidic bond is removed, hence the name debrancher.Unlike phosphorylase, which removes glucose moieties from glycogen in the form of glucose-1-phosphate, debrancher releases 1 free glucose moiety from each branch point. After the cleavage of the branch site, phosphorylase attacks unbranched portions of the glycogen molecule until the enzyme is stymied by the appearance of another branch point, at which point debranching enzyme once again is called into play. Eventually, large portions of the glycogen molecule are degraded to free glucose by the action of the amylo-alpha1,6-glucosidase activity of debranching enzyme and to glucose-1-phosphate by the action of phosphorylase.
Then, the transferase component of the debranching enzyme transfers the 3 glucose residues from the short branch to the end of an adjacent branch of the glycogen molecule. The glucosidase component of debranching enzyme then removes the glucose moiety remaining at the alpha1,6 branch point. In the process, the branch point formed by the alpha1,6-glucosidic bond is removed, hence the name debrancher.
Unlike phosphorylase, which removes glucose moieties from glycogen in the form of glucose-1-phosphate, debrancher releases 1 free glucose moiety from each branch point. After cleaving the branch site, phosphorylase attacks unbranched portions of the glycogen molecule until the enzyme is stymied by the appearance of another branch site, at which point debranching enzyme is once again called into play. Eventually, the entire glycogen molecule is degraded to free glucose by the action of the amylo-alpha1,6-glucosidase activity of debranching enzyme and to glucose-1-phosphate by the action of phosphorylase.
The glucose-1-phosphate may be used in various biosynthetic reactions, or it may be converted to glucose-6-phosphate by the action of phosphoglucomutase. The glucose-6-phosphatase so formed may be used for energy production via either the glycolytic or the hexose monophosphate pathway, or it may be converted to free glucose by the action of glucose-6-phosphatase.
Hypoglycemia
Hypoglycemia is the primary clinical manifestation of GSD III. At least in its early stages, hypoglycemia is caused by the defect in glycogenolysis that results from deficient activity of debranching enzyme. Because of deficient debrancher activity in GSD III, only a small portion of the glucose moieties stored in the liver as glycogen is readily available for glucose homeostasis. As a result, patients may experience significant hypoglycemia, even after a relatively short fast. [10] However, in contrast to patients with GSD I, gluconeogenesis is normal in patients with all forms of GSD III. This probably explains why the hypoglycemia observed in patients with GSD III is usually less severe than that routinely encountered in patients with GSD I. Nonetheless, be aware that patients with GSD III can experience hypoglycemia sufficiently severe to induce hypoglycemic seizures and to cause brain damage and even death.
Hepatic abnormalities
The mechanism responsible for the liver damage that occurs in GSD III is unknown. During infancy and early childhood, plasma transaminase levels are routinely elevated, often to very high levels. Although the recurrent bouts of hypoglycemia may cause hepatic damage, as has been suggested, hepatic fibrosis and cirrhosis do not occur in GSD I, in which the bouts of hypoglycemia are usually more frequent and more severe. Another hypothesis suggests that the abnormally structured glycogen may play a role in liver damage; however, no evidence supports this theory. Similarly, no explanation has been provided for the hepatic adenomas and hepatocellular carcinomas that occasionally develop in patients with GSD III. [11]
An entirely realistic goal, given modern treatment modalities, is reduction of the incidence of all hepatic complications by preventing bouts of hypoglycemia. Stabilizing blood glucose levels within the reference range significantly reduces incidence of hepatic adenomas and hepatocellular carcinoma in patients with GSD I. No theories explain why hepatic signs and symptoms of GSD III usually improve with advancing age and even may disappear after puberty.
Skeletal myopathy and cardiomyopathy
The mechanism causing the myopathy and the cardiomyopathy that occurs often in GSD IIIa is unknown, although this muscle damage is suggested to be attributed to recurrent bouts of hypoglycemia. However, myopathy and cardiomyopathy do not occur in GSD I, despite this condition's typically more frequent and severe bouts of hypoglycemia. Abnormally structured glycogen may play a role in the myopathy, but this hypothesis lacks support. [5, 12]
Miscellaneous abnormalities
During infancy and early childhood, patients with GSD III may have hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation, conditions similar to those in GSD I. Clinical presentation of the 2 diseases in children aged 3-8 years may be almost indistinguishable. Because all of the steps in glucose metabolism, including glucose-6-phosphatase and the transport system for glucose-6-phosphate, are intact in patients with GSD III, levels of phosphorylated glycolytic intermediates are not elevated. As a result, blood lactate and uric acid levels usually are within reference ranges, unlike the marked elevations that routinely occur in patients with GSD I. Nonetheless, modestly elevated blood levels of lactate and uric acid occasionally occur in patients with GSD III for no known reason. [10]
Similarly, patients with GSD III catabolize fatty acids normally because the activity of acetyl CoA carboxylase or levels of malonyl CoA do not significantly increase. As a result, the hypoglycemia of patients with GSD III is often accompanied by significant fasting ketosis, a combination that does not occur in patients with GSD I.
Causes
All forms of GSD III display autosomal recessive inheritance and are caused by various mutations at chromosome band 1p21. Because so many mutations in the debrancher gene have already been identified, most affected patients probably are compound heterozygotes rather than true homozygotes. [13, 14]
Patients with GSD IIIa apparently have a generalized debrancher activity deficiency, which has been identified in the liver, skeletal muscle, heart, erythrocytes, and cultured fibroblasts. Research has demonstrated that the progressive myopathy and/or the progressive cardiomyopathy develop only in patients with this generalized debrancher activity deficiency.
Patients with GSD IIIb are deficient in debrancher activity in the liver but have normal enzyme activity in muscle. The molecular biology of GSD IIIa and IIIb is an extremely active area of research; several quite different mutations, including different types of mutation, in the debrancher gene can produce GSD IIIa. GSD IIIb is caused by 2 different mutations in exon 3 at the amino acid codon 6. No known mechanism explains how these exon 3 mutations permit debranching enzyme activity in muscle but not in liver. [15]
Rare forms of GSD III
In addition to GSD IIIa and GSD IIIb, 2 relatively rare forms of the disease are termed GSD IIIc and GSD IIId. Only 1 or 2 cases of GSD IIIc are documented, and its clinical manifestations have not been fully described. GSD IIIc has intact debrancher transferase activity but deficient glucosidase activity. Because no patient with this form of GSD III has been reported in the past 20-30 years, this condition may have been a "family disease." The molecular biological basis for this rare form of GSD III is unknown.
A significant number of GSD IIId cases have been described. The condition is clinically indistinguishable from GSD IIIa. In GSD IIId, debrancher glucosidase activity is normal, but transferase activity is deficient in both liver and muscle tissues. [16]
Epidemiology
Frequency
United States
No reliable estimates of GSD incidence are available because of the lack of newborn screening programs for these disorders. The estimated incidence of GSD III in North America is approximately 1 case per 100,000 live births.
International
Based on several European studies, the overall GSD incidence is approximately 1 case per 20,000-25,000 live births. As GSD III accounts for approximately 24% of all GSD cases, its estimated incidence in Europe is approximately 1 case per 83,000 live births. Disease frequency is higher in certain populations, such as the Inuit population of North America and Sephardic Jewish people of North African descent, among whom the incidence is approximately 1:5,066 or 1:5,400, respectively. [17] This incidence for the homozygous condition translates to a frequency of 1:37 for the heterozygous state. All North African Jewish patients with GSD III have GSD IIIa. The highest known GSD III prevalence occurs in the Faroese population of the Faroe Islands, where the estimated incidence is approximately 1:3600 due to a founder effect. [18]
Mortality/Morbidity
Note that the clinical manifestations of GSD III, even within the various subtypes, vary dramatically from patient to patient. These differences are termed microheterogeneity. Although the basis for microheterogeneity in GSD III is not understood, the level of residual debrancher activity does not determine clinical severity. An example of this microheterogeneity occurs in the families of North African Jewish patients with GSD IIIa whose peripheral neuromuscular impairments vary from minimal to severe, yet all have both liver and muscle involvement and precisely the same single mutation: the deletion of T at position 4455 (4455delT) in both alleles. [19]
Hypoglycemic symptoms and complications are frequent and nonspecific and include the following:
-
Irritability
-
Tantrums
-
Tremulousness
-
Poor feeding
-
Respiratory distress
-
Apnea
-
Bradycardia
-
Lethargy
-
Confusion or apathy
-
Twitching or shakiness
-
Seizures
-
Inappropriate behavior (eg, laughter, crying)
-
Poor concentration
-
Hypothermia
-
Headache
-
Pallor
-
Palpitations
-
Hypotonia
-
Sweating
-
Coma
-
Sudden death
Long-term complications of hypoglycemia and/or storage of abnormal glycogen include the following:
-
Cirrhosis
-
Liver failure
-
Hepatic adenomas
-
Hepatocellular carcinomas
-
Exercise intolerance
-
Muscle wasting and weakness
-
Cardiomegaly
-
Renal failure secondary to myoglobinuria (rare)
-
Dilated hypertrophic cardiomyopathy with ventricular dysfunction [20]
Race
GSD III has been reported in several racial and ethnic groups, including white Europeans, Africans, Hispanics, Jews, Aboriginal North Americans, and Asians. GSD III is especially frequent among Sephardic Jewish people from North Africa; all affected people in this group have GSD IIIa.
Sex
All forms of GSD III occur with equal frequency in both sexes because the disorder has autosomal recessive inheritance.
Age
GSD III is an inborn error of metabolism; the condition is present from the moment of conception. Age of first clinical appearance varies dramatically from patient to patient. Hypoglycemia is rare in neonates but often manifests at age 3-4 months, an age when many parents reduce feeding frequency. Hepatic symptoms may be so mild that the diagnosis is not confirmed until adulthood, when the patient first manifests signs and symptoms of neuromuscular disease.
Prognosis
Patients receiving proper management of their blood glucose levels should not encounter life-threatening hypoglycemia. Evidence has shown that most primary manifestations can be avoided or reduced if euglycemia can be maintained. [21, 22]
Some patients still develop cirrhosis and even liver failure. Liver transplantation is an option. [23]
Cirrhosis may lead to hepatocellular carcinoma.
Some patients develop hypertrophic cardiomyopathy, yet overt cardiac dysfunction is rare.
Patient Education
Teach all caregivers and sufficiently mature patients how to recognize signs of impending hypoglycemia.
Teach all caregivers and sufficiently mature patients how to manage hypoglycemic episodes.
Teach all caregivers and sufficiently mature patients how to measure blood glucose levels.
Teach all caregivers how to insert an nasogastric tube (NGT) and how to use an infusion pump.
Provide intensive nutritional education to caregivers and sufficiently mature patients.
Encourage sufficiently mature patients to participate in the dietary management of their disease.
The following organizations provide excellent information for families of patients with GSD III:
-
Association for Glycogen Storage Disease; Durant, Iowa; 563-785-6038
-
Schematic illustration of the degradation of glycogen by the concerted action of the enzymes phosphorylase and debranching enzyme. First, phosphorylase removes glucose moieties (linked to their neighbors via alpha1,4 glucosidic bonds and depicted as the 7 black circles) from the unbranched outer portions of the glycogen molecule until only 4 glucosyl units (depicted as the 3 green circles and the 1 red circle) remain before an alpha1,6 branch point. The transferase component of debranching enzyme then transfers the 3 (green) glucose residues from the short branch to the end of an adjacent branch of the glycogen molecule. The glucosidase component of debranching enzyme then removes the glucose moiety (depicted as the red circle) remaining at the alpha1,6 branch point. In the process, the branch point formed by the alpha1,6 glucosidic bond is removed, hence the name debrancher.Unlike phosphorylase, which removes glucose moieties from glycogen in the form of glucose-1-phosphate, debrancher releases 1 free glucose moiety from each branch point. After the cleavage of the branch site, phosphorylase attacks unbranched portions of the glycogen molecule until the enzyme is stymied by the appearance of another branch point, at which point debranching enzyme once again is called into play. Eventually, large portions of the glycogen molecule are degraded to free glucose by the action of the amylo-alpha1,6-glucosidase activity of debranching enzyme and to glucose-1-phosphate by the action of phosphorylase.
-
Schematic representation of a portion of a molecule of glycogen. Open circles represent the glucose moieties connected to each other via alpha1,4 linkages. Solid circles represent the glucose moieties connected to their neighbors via alpha1,6 linkages. Thus, each solid circle represents a branch point in the molecule.








