Introduction And Epidemiology
Background
The GM2 gangliosidoses are a group of lysosomal lipid storage disorders caused by mutations in at least 1 of 3 recessive genes: HEXA, HEXB, and GM2A. Normal products of all 3 genes are required for normal catabolism of the GM2 ganglioside substrate. Deficient activity of these enzymes leads to accumulation of the substrate inside neuronal lysosomes, leading to cell death. The products of the 3 genes are, respectively, the alpha subunits of b-hexosaminidase A (Hex A; EC 3.2.1.52), the beta subunits of Hex A (EC 3.2.1.52), and the GM2 activator protein. Hydrolysis of GM2 ganglioside requires a normal GM2 ganglioside–GM2 activator–Hex A complex. [1]
Hex A is a dimer and has the structure alpha-beta. The alpha subunit is encoded by the HEXA gene at band 15q23-q24; the beta subunit is encoded by the HEXB gene at band 5q13. A site on the alpha subunit acts against negatively charged sulfated substrates, while a site on the beta subunit acts against neutral water-soluble substrates.
β-Hexosaminidase B (Hex B) is a dimer of beta chains. It hydrolyzes GM2 and its neutral asialo derivative GA2. Both subunit precursors acquire the mannose 6-phosphate marker for recognition by lysosomes.
Hexosaminidase S (Hex S) is a dimer of alpha chains; it is a normal constituent of plasma and degrades a wide range of glycoconjugates containing β-linked N -acetylhexosaminyl residues.
Lysosomal storage disorders (LSDs) are caused by a genetic deficiency in the enzymatic function of the hydrolases stored within lysosomes, causing an accumulation of materials meant for degradation. There are a few mechanisms by which a genetic defect can lead to an LSD. In the case of GM2 gangliosidosis, an activator protein is deficient, which leads to the accumulation of ganglioside GM2 and related glycolipids in neuronal cells. [2]
Disease classification
Tay-Sachs disease (TSD) and its variants are caused by absence or defects of the alpha subunit of Hex A. [3]
-
Type I GM2 gangliosidosis is also known as classic infantile acute TSD, B variant, pseudo-AB variant, or Hex A deficiency.
-
Type III GM2 gangliosidosis is also known as juvenile subacute TSD.
-
The B1 variant of GM2 gangliosidosis is also known as late infantile subacute-to-chronic TSD; it is characterized by a defect in formation and stabilization of the alpha subunit active site.
-
GM2 gangliosidosis, also known as adult chronic-type TSD is characterized by a pseudodeficiency mutation in one or both HEXA alleles.
Sandhoff disease (SD) and its variants are caused by absence or defects of the beta subunit of Hex A and the subunits of Hex B. Type II GM2 gangliosidosis is also known as SD (also including the juvenile subacute type); Hex A and Hex B deficiency; or GM2 gangliosidosis, O variant.
Hexosaminidase activator deficiency is caused by absence or defects of the hexosaminidase activator. Type AB GM2 gangliosidosis is also known as hexosaminidase activator deficiency.
Table 1. Genetic Characteristics of GM2 Gangliosidoses (Open Table in a new window)
Gene Features |
HEXA |
HEXB |
GM2A |
Chromosome location |
Band 15q23-q24 |
Band 5q13 |
Band 5q31.3-q33.1 |
Product |
Alpha subunit of Hex A; subunits of Hex S |
Beta subunit of Hex A; subunits of Hex B |
GM2 activator protein |
Heat sensitivity, pH |
Heat labile, acidic |
Heat stabile, basic |
Heat stabile, acidic |
TSD-B variant TSD-pseudo-AB variant classic infantile acute TSD |
Most severe phenotype; both alleles absent or mutated (deficient Hex A; may lead to increased levels of Hex B) |
Normal |
Normal |
B1 variant TSD |
Mutated (near-normal Hex A is inactive toward GM2) |
Normal |
Normal |
Adult chronic-type TSD |
Mutated, pseudodeficiency mutation in at least 1 allele |
Normal |
Normal |
SD, O variant |
Normal |
Both alleles absent or mutated (deficient Hex B; may lead to some Hex S activity) |
Normal |
Hexosaminidase Paris (SD) |
Normal |
At least 1 mutated allele (some normal Hex B activity) |
Normal |
AB variant (hexosaminidase activator deficiency) |
Normal (increased amounts of product) |
Normal (increased amounts of product) |
Absent or mutated |
Known mutations |
105 |
31 |
5 |
Pathophysiology
Clinical symptoms of progressive neurodegeneration and developmental delay are caused by accumulation of GM2 ganglioside in lysosomes, forming membranous cytoplasmic bodies (MCBs) in neuronal cells. [4] MCBs also contain cholesterol and phospholipid. Accumulation of storage material leads to unscheduled cell death. The classic infantile forms of the 3 subgroups of GM2 gangliosidosis (ie, TSD, SD, and hexosaminidase activator deficiency) have their onset in infancy and lead to death by age 4 years. The severity of the disease correlates inversely with the amount of residual Hex A activity in lysosomes. Usually, but not always, later disease onset corresponds with slower disease progression.
Patients with Hex A and Hex S deficiency (variant B) were found to have accumulated GM2 and minor amounts of GA2 in their brains. One patient without Hex deficiency (AB variant) had considerable GA2 levels and even higher GM2 levels. Patients with Hex A and Hex B deficiency (O variant) contained the highest GA2 levels and lowest GM2 levels.
Patients with nonclassic variants of GM2 gangliosidoses may present with other symptoms, such as ataxia, dystonia, psychosis, and/or muscle wasting. The various symptoms are not pathognomonic and may occur in many other conditions, including conditions caused by exposures to certain environmental agents.
Frequency
See the list below:
-
United States
TSD
Before community-based carrier screening programs were developed for the Ashkenazi Jewish population, [5] 1 per 2500-3600 newborns of Ashkenazi Jewish descent was affected. Heterozygote screening and counseling programs led to a 90% reduction of the disease in the high-risk Jewish population.
One in 25-30 persons of Ashkenazi Jewish descent is a heterozygous unaffected carrier of a HEXA mutation.
In certain isolated populations, such as Louisiana Cajuns and Pennsylvania Dutch, the prevalence of TSD is as high as or higher than in individuals of Ashkenazi Jewish descent.
In the general population, 1 per 320,000 newborns is affected.
One in 283 individuals is a heterozygous unaffected carrier of a HEXA mutation.
SD
One per 309,000 non-Jewish newborns is affected.
One in 278 persons of non-Jewish descent is a heterozygous unaffected carrier of a HEXB mutation.
One in 7 persons of the Maronite community in Cyprus is a carrier.
Approximately 1 per 1 million Jewish newborns is affected.
Approximately 1 per 500 Jewish persons is a heterozygous unaffected carrier of a HEXB mutation.
Hexosaminidase activator deficiency: This condition is extremely rare.
-
Internationally
TSD
One per 360,000 newborns in the general population is affected.
One in 280 individuals is a heterozygous unaffected carrier of a HEXA mutation.
The incidence in French Canadians of the eastern St. Lawrence River Valley area of Quebec is similar to that in persons of Ashkenazi Jewish descent.
In the Cordoba region of Argentina, among Creole, Spanish, and native peoples, 1 in 26 individuals is a carrier.
Increased frequencies were reported in Moroccan Jews (1 in 110 is a carrier) as well as in some isolated populations in Switzerland and Japan.
One in 140 Iraqi Jews is a carrier.
In Saudi Arabia, the cause is private mutations, which prevents replicating the successful approach used with Ashkenazi Jewish population. Therefore, alternative solutions must be implemented for other autosomal recessive diseases. [6]
SD
Approximately 1 per 310,000 non-Jewish newborns is affected.
Approximately 1 per 1 million Jewish newborns is affected.
Increased incidences were reported in Creoles of northern Argentina, Metis Indians of northern Saskatchewan, individuals of Lebanese heritage, and Hispanics of Mexican or Central American heritage. Four patients from different families were detected in Cyprus.
Hexosaminidase activator deficiency: This condition is extremely rare.
Mortality and morbidity
See the list below:
-
TSD
The classic infantile form is usually fatal by age 2-4 years.
In the late infantile subacute, B1 variant form, disease progression is particularly aggressive, leading to death within 2-4 years of disease onset.
In the juvenile subacute form, death occurs by age 10-15 years; it is usually due to infection and is preceded by several years in a vegetative state with decerebrate rigidity.
In the adult chronic form, most patients survive to age 60-80 years.
-
SD: Death generally occurs by age 4 years.
-
Hexosaminidase activator deficiency: The 5 cases reported involved the infantile acute form of TSD. All 5 infants known to be affected with hexosaminidase activator deficiency died in infancy.
Ethnicity
See the list below:
-
TSD
In the past, 99% of affected individuals were of Ashkenazi Jewish descent; yet, parental consanguinity was relatively infrequent. As a result of targeted community-based carrier screening, a relatively greater proportion of cases of TSD are now observed in non-Jewish individuals. Sephardic Jews have no increased risk.
In certain isolated populations, such as French Canadians of the eastern St Lawrence River Valley area of Quebec, Louisiana Cajuns, and Pennsylvania Dutch, the incidence of TSD is as high as or higher than in Ashkenazi Jews. Moroccan Jews appear to have an increased incidence. Among Creole, Spanish, and native peoples of the Cordoba region of Argentina, 1 in 26 individuals is a carrier. In non-Ashkenazi Jewish patients with TSD, parental consanguinity is frequent.
The disease has been found in blacks and in Asians. However, no cases have been reported in Eskimo, Gypsy, or Mongolian populations.
-
SD: Although the disease is panethnic, increased prevalences were reported in Creoles of northern Argentina, Metis Indians of northern Saskatchewan, individuals of Lebanese heritage, and Hispanic persons of Mexican or Central American heritage. Several affected were Maronites of Greek-Cypriot heritage.
-
Hexosaminidase activator deficiency: The 5 affected individuals reported were Indian, Saudi Arabian, Spanish, black, and Laotian.
Sex
See the list below:
-
In all GM2 gangliosidosis subgroups, males and females appear to be equally affected.
Age
See the list below:
-
TSD
Classic infantile acute form - Onset during age 4-8 months
Late infantile and juvenile subacute forms - Onset from age 2-10 years
Adult chronic form - Onset from childhood to adulthood
Late-onset form - Onset during adulthood
-
SD
Infantile acute form - Onset within the first 6 months of life
Late-onset subacute form - Onset during age 2-10 years
Adult chronic form - Onset in adulthood
-
Hexosaminidase activator deficiency: All 5 reported individuals exhibited symptoms in infancy.
Tay-Sachs Disease - GM2 Gangliosidosis Type I, Type III, Chronic, And B1 Variant
TSD was described by Warren Tay in 1881 and by Bernard Sachs in 1887. The disease and its variants are caused by an absence or deficiency in the alpha subunit of Hex A. The severity of the disease inversely correlates with the level of Hex A activity. A deficiency in the GM2 A activator protein also results in symptoms of TSD because normal GM2 ganglioside degradation requires binding of the ganglioside to a GM2 activator–Hex A complex. [7]
Type I GM2 gangliosidosis - Classic infantile acute Tay-Sachs disease
A total lack of Hex A results in defective lysosomal degradation of the GM2 ganglioside. Patients have 0.1% of normal Hex A enzyme activity. The omnipresent accumulation of GM2 gangliosides/MCBs in neuronal cells leads to progressive neurodegeneration.
Children with type I gangliosidosis do not appear to be affected at birth. Loss of milestones occurs in infancy. Neurologic symptoms as well as muscular weakness, which leads to paralysis, start by age 3-5 months. Most patients never walk and exhibit increasing apathy and inattention. Hyperacusis (ie, persistent extension response to sound, startle reaction) can aid in early diagnosis. After age 8-10 months, deterioration is rapid as neurodegeneration progresses. Macrocephaly due to reactive cerebral gliosis typically begins at age 18 months. Eventually, patients have difficulty swallowing, uncontrolled seizures, spasticity, blindness, and dementia. Death from bronchopneumonia usually occurs by age 4 years. Identical symptoms occur in type AB GM2 gangliosidosis (ie, hexosaminidase activator deficiency). [8]
Electroretinographic and EEG findings are normal, but visual-evoked responses are abnormal. Lipid-laden ganglion cells appear as a gray-white area around the retinal fovea centralis (a central cherry-red spot with a halo or a perifoveal white patch) that can be detected by means of funduscopy. Ballooned neurons are present throughout the CNS.
Clinical variation is noted. Specific mutations may cause abnormalities such as muscle atrophy (beginning distally), pes cavus, foot drop, spasticity, mild ataxia of the limbs and trunk, dystonia, and dysarthria. [9] Intellectual, visual, and hearing function may remain normal, and seizures may be absent. Progressive proximal muscle wasting, leg weakness, and fasciculations may be noted. Electromyographic abnormalities may be noted, and creatine phosphokinase isoenzyme levels may be elevated. Muscle biopsy may suggest anterior horn disease. Nine patients from 4 unrelated Ashkenazi Jewish families with a variant form of Hex A deficiency presented with atypical Friedreich ataxia.
The abnormal phenotypes have been postulated to be due to one mutation in the HEXA gene and another one in a gene not yet identified or, alternatively, from one classic mutation and one mild mutation. [10]
B1 variant (pseudo–AB variant) gangliosidosis - Late infantile Tay-Sachs disease
Hex A levels are nearly normal, but hydrolysis of GM2 ganglioside and the sulfated Hex A substrate 4-methylumbelliferyl-beta-D-N -acetylglucosamine-6-sufate are virtually absent because of a mutation in the alpha subunit that leads to defective folding and instability of the catalytic domain. A patient usually has one absent or defective HEXA allele and one allele that allows some HEXA expression. Dementia or chronic organic brain syndrome may occur. Neuronal MCB storage is milder than in the infantile disease; however, disease progression is particularly aggressive, leading to death within 2-4 years of onset.
One Puerto Rican girl started losing mental capacity at age 6 and had seizures at age 8, cerebellar symptoms at age 9, pyramidal symptoms and dystonia at age 10, and dysphagia at age 14. She died at age 14 1/2 years.
In infantile and adult TSD, the glycolipid storage occurs mostly in the hippocampus, brain stem, spinal cord, and cerebellum, while the cortex may remain uninvolved.
Type III GM2 gangliosidosis - Juvenile subacute Tay-Sachs disease
Enzyme activity is 0.5% of normal activity. The residual Hex A enzyme activity can be diagnosed at age 3-10 years. Affected children experience deterioration of gait and posture, which begins early but progresses slowly. Progressive deterioration in speech and life skills may occur. Cognition may decline, although normal intelligence has also been reported. Dementia or chronic organic mental syndrome and/or seizures may occur. Patients may experience increased spasticity. Vision may be normal or may decline late, and optic fundi may be absent. Optic atrophy and retinitis pigmentosum may occur late. Cerebellar atrophy occurs in approximately 50% of affected individuals; neuropathy and mental disorders may occur. Affected individuals survive into late childhood or adolescence. Death is usually by age 15 years because of infection and is preceded by several years in a vegetative state with decerebrate rigidity.
Chronic GM2 gangliosidosis - Adult Tay-Sachs disease
Enzyme activity is less than 10% and is usually 2-4% of normal activity. TSD is usually diagnosed when the patient is in early adolescence, although some psychomotor regression may begin in early childhood.
Hex A–deficient adults are usually compound heterozygotes, in which one allele has a G269S mutation and the other allele a mutation for infantile TSD. Patients who have a G269S mutation on each allele (ie, homozygotes) are only mildly affected.
Neurologic defects progress slowly and may lead to lack of coordination; hand tremors; progressive dystonia; dysarthria, dyskinesia; choreoathetosis; ataxia; spinocerebellar degeneration; motor neuron disease with proximal muscle wasting, cramping, and weakness; and/or fasciculations. More than one third of affected individuals present with psychosis. Psychotic patients should not be treated with antidepressants because this further depletes Hex A levels. Psychiatric abnormalities include acute hebephrenic schizophrenia, agitation, delusions, hallucinations, paranoia, and recurrent depression that may precede neurologic defects, but dementia is usually not prominent.
Some patients may present with symptoms suggesting spinocerebellar degeneration, Friedreich ataxia, amyotrophic lateral sclerosis, or spinal muscular atrophy. The patient's intellect may be either mildly impaired or within normal limits. Clinical phenotypes vary even within a family. Rectal ganglion cells show ballooning and onion skin cytoplasmic bodies/MCBs.
Late-onset Tay-Sachs disease
In very rare cases, adult-onset TSD has been reported. Patients with this subtype may experience life-long subtle clumsiness, as well as athletic difficulties, which develop into a rare mimicry condition of amyotrophic lateral sclerosis. In one case, a 35-year-old Hungarian man with no known Jewish ancestry was diagnosed with late-onset TSD following genetic testing, which identified a heterogeneous pathogenic mutation with both 1499T deletion and an 805G>A substitution in the HEXA gene. [11]
Clinically asymptomatic individuals
Because of considerable heterogeneity in expression of mutations, homozygous mutants or compound heterozygotes (with 11-20% of normal enzyme activity) do not have symptoms of TSD.
Genetics
Disease genetics are as follows:
-
Mode of inheritance - Single gene, autosomal recessive
-
Gene and chromosomal locus -HEXA at band 15q23-q24
-
Gene product - Alpha subunit of Hex A
The gene is about 35 kilobases (kb) long, and the coding region consists of 14 exons.
Mutations in codons 170−211 are the most likely to interfere with enzyme activity.
The two genes, HEXA and HEXB, share approximately 55% homology at the nucleotide and amino acid levels.
The amino acid Arg 178 in the active site of the a chain corresponds to Arg 211 in the active site of the b chain.
Table 2. Some Mutations in the HEXA Gene* (Open Table in a new window)
Group |
HEXA Mutation |
Frequency in Carriers |
Type |
Ashkenazi Jews |
1278+TATC, exon 11 |
75-80% |
Death in infancy |
|
IVS12+1G>C, intron 12 splice site mutation leads to a 35-bp deletion |
15-18% |
Death in infancy |
|
G269S, exon 7 |
3% |
Adult onset, variable phenotypes (homozygotes or compound heterozygotes, usually with 1278+TATC or IVS12+1G>S) |
|
R247W or R249W, exon 7 |
2% |
Benign pseudodeficiency |
Non-Ashkenazi Jews |
delF304, exon 8 |
Moroccan Jews, French, Italian, Portuguese (percentage not known) |
Death in infancy |
Non-Jewish enzyme- deficient TSD carriers |
1278+TATC, exon 11 |
5% in French Canadians and Acadians of Louisiana and 20% of non-Jewish European-derived populations |
Death in infancy |
|
5'UTRdel, 7.6-kb deletion (no mRNA) |
80% in non-Jewish French Canadians from eastern Quebec |
Death in infancy (first TSD mutation discovered) |
|
IVS9+1G>A, intron 9 splice site mutation |
15% in northern Europeans (French, Celtic), Cajun, and Pennsylvania Dutch |
Death in infancy |
|
IVS12+1G>C, intron 12 |
< 1% |
Death in infancy |
|
IVS5-1G>T |
Japanese most affected (no known percentage) |
Death in infancy |
|
12-bp del, exon 10 |
Turkish |
Death in infancy |
|
G454D, exon 12 |
Turkish |
Death in infancy |
|
R178H, exon 5 |
Northern Portuguese and unrelated Europeans in Mediterranean region |
B1 variant, severe late infantile (heterozygous with mRNA negative mutation or severe juvenile (homoallelic) |
|
R178L, exon 5 |
English |
Between infantile and B1 variant |
|
R178C, exon 5 |
Czechoslovakian |
B1 variant |
|
G250D, exon 7 |
Lebanese |
Juvenile |
|
D258H, exon 7 |
Scottish-Irish |
B1 variant |
|
R499H, exon 13 |
Mixed Jewish/Scottish-Irish |
Juvenile, early onset, rapidly progressive |
|
R504H, exon 13 |
Assyrian, Armenian, Lebanese, East European |
Juvenile |
|
G269S, exon 7 |
5% in Americans and Europeans |
Adult onset, variable phenotypes (homozygotes or compound heterozygotes with infantile TDS mutation) |
|
R247W or R249W, exon 7 |
35% |
Benign pseudodeficiency |
|
G250S |
80% in French Canadians |
Adult onset |
|
W474C |
|
Juvenile, later onset |
*More than 100 HEXA mutations spanning all 14 exons of the gene have been reported; approximately 70% cause acute TSD, approximately 20% cause subacute TSD, and approximately 9% cause chronic TSD. Approximately 58% of mutations are missense or nonsense mutations. Some genotype-phenotype correlations are described in Online Mendelian Inheritance in Man (OMIM). See also The Human Gene Mutation Database, HEXA. |
|||
As many as 10% of carriers confirmed by DNA analysis may have serum Hex A levels in the noncarrier range because of the presence of 2 HEXB polymorphisms: 619A>G and delTG. Further testing with leukocyte Hex A profiles is recommended.
Workup and physical examination
Both homozygotes and heterozygotes have a reduced concentration of sphingomyelin in their RBCs.
Affected individuals have deficient Hex A activity and normal levels of the Hex B isozyme. Hexosaminidase levels can be tested in serum, white blood cells, cultured somatic tissues including amniocytes, and even in dried blot spots on filter paper. Hex A is heat labile, whereas Hex B is heat stabile. Absence of the enzyme is confirmed by testing white blood cells, amniotic fluid, or a chorionic villus sample. Serum samples are used to test males and females who are not pregnant and not using oral contraceptives. White blood cells samples are used to test pregnant females, females using oral contraceptives, and individuals in whom the serum Hex A enzymatic level was in an inconclusive range. One half of carriers can be detected by enzyme assay screening. Abnormal results should be followed by DNA analysis to detect the disease-causing mutation, to exclude the presence of a pseudodeficiency allele, or both.
Most testing is performed using a synthetic substrate (4-methylumbelliferyl-b-D-N -acetylglucosaminide 6-sulfate [4-MUGS]), which can only be cleaved by the catalytic site on the a subunit of Hex. In activator-deficient AB variant TSD, testing is performed with labeled GM2 ganglioside not with a chromogenic substrate. Also, two pseudodeficiency alleles (R247W or R249W) result in reduced enzyme activity when tested with 4-MUGS, but they do not lead to disease in the tested individual because the normal GM2 ganglioside substrate is processed normally. A pseudoallele was found in 2% of Jewish patients and in 35% of non-Jewish patients who were determined to be heterozygous carriers by means of enzyme testing. Individuals who have a pseudodeficiency HEXA mutation are not at risk of having a child with GM2 gangliosidosis.
Biochemical testing is recommended in low-risk populations because it detects a higher percentage of carriers, whereas DNA analysis detects less than 50% of carriers in these populations.
In 1970, TSD became the prototype for genetic disease prevention. North American individuals with a family history positive for a GM2 gangliosidosis or Ashkenazi Jewish heritage agreed to be tested. To date, more than 1.3 million adults have been screened. Among them, 1350 couples were carriers. The screening programs and monitoring of at-risk pregnancies have reduced the prevalence of TSD in this population by more than 90%. Screening of at-risk individuals is recommended in guidelines of the American College of Medical Genetics and of the American College of Obstetricians and Gynecologists. [12]
DNA diagnostics
A DNA diagnostic evaluation is recommended (1) to differentiate pseudodeficiency alleles from disease-causing alleles, (2) to identify disease-causing alleles in affected individuals, and (3) to identify the disease-causing mutation in an affected individual's parents, who are both obligate carriers. At least one laboratory successfully performed single-cell testing on 95% of 248 samples; only 7 of the samples had to be excluded from further analysis.
Identification of the mutation can lead to identification of other carriers in the family and to evaluation of childbearing options or prenatal diagnosis. Testing, genetic counseling, and prenatal diagnosis are available (for more information, see Gene Tests). Pre-embryonic genetic diagnosis may be available for specific mutations.
In Ashkenazi Jews, direct detection of 3 mutations has 96-98% sensitivity, and biochemical analysis has 98% sensitivity. In non-Jewish North Americans, more than 20 mutations have been found; 35% have a benign 739C>T or 745C>T (R247W or R249W) mutation.
Ophthalmoscopic examination
The cherry-red central spot or perifoveal white patch seen in the retinal fovea centralis of the retinal macula of those affected with infantile and juvenile TSD is not seen in those affected with chronic TSD. The image below is an example of retina changes seen in TSD.
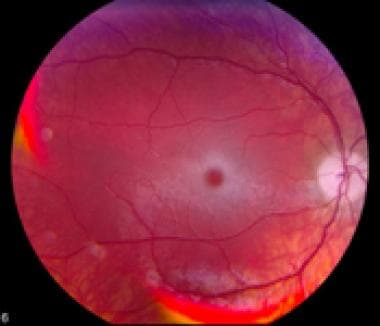 Fundus photograph showing retina changes associated with GM2 gangliosidosis.
Fundus photograph showing retina changes associated with GM2 gangliosidosis.
Evaluate for hyperreflexia with sustained ankle clonus during the physical examination.
-
Histologic studies: In infantile cases, MCBs stain strongly with periodic acid-Schiff (PAS) stain on frozen sections but not on paraffin sections. Electron microscopic examination of biopsied rectal cells reveals MCB accumulation that leads to enlarged ganglion cells. Muscle biopsy reveals denervation atrophy.
-
Imaging studies: CT scanning or MRI may be used to detect cerebellar atrophy.
-
Electromyography: Denervation and reinnervation may be seen in the adult chronic form.
Medical care
Enzyme replacement has not yet been successful. [13] Because no specific treatment is available for TSD, treatment is directed at the symptoms and major associated conditions. Treatment is supportive and aimed at providing adequate nutrition and hydration. The airway must be protected. Seizures can be controlled initially with conventional anticonvulsant medications, but the progressive nature of the disease may require alteration of dosage or medication. Infectious diseases should be managed. In advanced disease, good bowel movement should be maintained and severe constipation should be avoided.
A clinical trial to investigate the pharmacokinetics and tolerability of miglustat (Zavesca) in infantile and juvenile GM2 gangliosidosis is currently underway. When investigated in late-onset TSD, miglustat was not shown to yield measurable benefits.
Some medications were associated with severe neurological symptoms, for example haloperidol, risperidone, and chlorpromazine.
Contacting national support groups may be helpful (see Further Information).
Consulting a medical geneticist is essential. Furthermore, evaluation by a psychiatrist is recommended. In adult-onset TSD, the response to tricyclic antidepressants and phenothiazines may be unexpected or poor; these drugs inhibited Hex A activity in vitro and induced lysosomal lipidosis in fibroblasts and accumulation of lipids in experimental animals in vivo. Lithium salts and electroconvulsive therapy were reported to be beneficial in ameliorating episodes of clinical depression, at least for some time.
Sandhoff Disease - GM2 Gangliosidosis Type II
Sandhoff and colleagues initially described this disorder in 1968 and 1971. SD and its variants are caused by defects in the HEXB gene, which codes for the beta subunit of Hex A and the subunits of Hex B. SD is also known as hexosaminidase A and B deficiency and GM2 gangliosidosis, O variant. In SD, the absence of or deficiency in Hex A causes defective lysosomal degradation and accumulation of secondary lysosomes. The absence of or deficiency in Hex B causes defective degradation of neutral substrates and dermatan sulfate, leading to accumulation of globosides in the serum, spleen, liver, lymph nodes, lungs, kidneys, and erythrocytes.
Because patients also lack or have defective Hex A activity, they exhibit symptoms of TSD and physical and visceral organ changes. Unlike TSD, most, if not all, cases occur in persons who are not of Ashkenazi Jewish descent. Patients have enzyme levels that are 5% of normal levels.
As in TSD, patients develop muscle weakness within the first 6 months of life, with startle reaction, blindness, cherry-red spots, macrocephaly, and progressive mental and motor deterioration. They may have a doll-like face. Nonneurologic symptoms may include organomegaly, skeletal abnormalities, and/or oligosacchariduria. Death usually occurs by age 4 years.
In one case, signs of heart involvement preceded those of nervous system disorder. A pansystolic murmur and cardiomegaly were found at age 3 months, and neurologic deterioration occurred at 8 months. Unlike patients with TSD, those with SD may present with coarse facies, macroglossia, megaloencephaly, cardiomegaly, hepatosplenomegaly, and high lumbar gibbus.
In one case, a black male had 20-24% of normal enzyme activity in blood but less than 2% in the liver. Three cases were reported in individuals from northern Saskatchewan, 7 in persons from Lebanon, and 36 in Creoles from Argentina.
A rare juvenile form of SD may result from an absence of normal Hex A levels but an increase in Hex B levels. Onset is at age 2-10 years. Patients have slowly progressing spinocerebellar disease with motor neuron involvement, dysarthria, muscle wasting, fasciculations, pyramidal dysfunction, and macular cherry spots. A rectal biopsy sample has shown MCBs in submucosal ganglion cells. Neuropathy and mental disorders are more prevalent in juvenile SD than in juvenile TSD.
Adult-onset SD (< 10% of normal enzyme activity) may begin in adolescence with slowly progressing motor neuron disease. Mutations appear to interfere with correct dimer formation.
Genetics
Disease genetics are as follows:
-
Single gene, autosomal recessive
-
Gene and chromosomal location -HEXB at band 5q13
-
Gene product of HEXB - Beta subunit of Hex A and subunits of Hex B
The gene is 40-45 kb long, and, like HEXA, the coding region comprises 14 exons.
The two genes, HEXA and HEXB, share approximately 55% homology at the nucleotide and the amino acid levels.
The amino acid Arg 211 of the b chain is the essential residue in the active site of HEXB; it corresponds to Arg 178 in the active site of the α chain.
Gln 355 is the essential residue at the substrate-binding site of the β chain.
-
The absence of beta subunits results in increased polymerization of alpha units to form Hex S
Table 3. Some Mutations in the HEXB Gene* (Open Table in a new window)
HEXB Mutation |
Frequency in Carriers |
Type |
16-kb deletion (5' end to IVS5, reported as 50-kb deletion by some investigators) |
30% in French or French Canadians |
All types of SD |
50-kb deletion (5' end to IVS6) |
... |
Infantile |
Del76A frameshift mutation, exon 1 |
84% of subtype, Greek-Cypriot Maronite |
Infantile |
S62L, exon 1, and partial deletion compound |
... |
Infantile |
IVS2+1G>A |
Cordoba region of Argentina in mixed Creole, Spanish, and native peoples |
Infantile |
IVS8+5G>C splice site mutation |
Greek-Cypriot |
Infantile |
P417L, exon 11 |
Japanese, Italian, French Canadians |
Juvenile and some infantile (in compound with S255R) |
Y456S, exon 11 |
... |
Juvenile in compound heterozygote |
-26IVS12 24-bp insertion |
... |
Paris†, juvenile or asymptomatic, latter likely due to a second mutation |
-16IVS13 18-bp insertion, splice site mutation |
French |
Paris†, juvenile to asymptomatic |
P417L, exon 11, and 16/50-kb deletion compound |
Japanese, Italian, French Canadian |
Adult |
P504S, 16/50-kb deletion compound |
... |
Adult |
R505Q, exon 13, and 16/50-kb deletion compound (heat labile) |
... |
Adult |
R533H, exon 13 |
Japanese |
Adult (in compound with IVS2+1G>A) |
*Of 31 mutations, approximately 65% caused acute SD, approximately 17% caused subacute SD, approximately 13% caused chronic SD, and approximately 4% caused benign SD. Of the mutations, 42% were missense or nonsense mutations. † Hex A plus/Hex B minus was originally called Hexosaminidase Paris. See also Online Mendelian Inheritance in Man (OMIM) and The Human Gene Mutation Database, HEXB. |
||
Workup
Enzyme assay: Hex B is heat stabile; however, certain benign mutations in the beta subunit can render Hex B heat labile and lead to inaccurate carrier testing and inaccurate prenatal diagnosis. The mutations are A543T and P504S, and one is unidentified. Carriers detected by means of enzyme assay should have a follow-up DNA diagnostic evaluation to identify the mutation.
Systemic organs have additional accumulation of sphingoglycolipids with a terminal hexosamine residue and undigested glycoprotein fragments. PAS-positive materials are seen in Kupffer cells in the liver; histiocytes in the spleen, lymph nodes, and lungs; and renal tubular epithelium on frozen section. The PAS-positive staining of systemic tissues is not seen in other forms of GM2 gangliosidosis and can be used for the differential diagnosis of SD.
Medical care
As in TSD, no treatment is available. Specific symptoms should be managed. Pre-embryonic genetic diagnosis may be available for certain mutations.
Consultation
Referral to a medical geneticist is essential.
Hexosaminidase Activator Deficiency, GM2 Gangliosidosis, Type A B
The GM2 activator is a water-soluble, glycosphingolipid-binding protein of low molecular weight that binds GM2 and GA2. It extracts these lipids from the membrane and presents them to Hex A. The active site on the α subunit of Hex A cleaves/solubilizes the lipids. Mutations in the GM2A gene render the GM2 activator incapable of binding GM2 or GA2 and lifting it out of the membrane, owing to defective hydrolysis.
The phenotype is that of classic infantile acute TSD. However, cerebral cortical neurons, in addition to MCBs, have zebra bodies and heterogeneous inclusions in astrocytes, oligodendrocytes, and microglia. Visceral organs are not involved.
Genetics
Disease genetics are as follows:
-
Single gene, autosomal recessive
-
Gene symbol and chromosome location -GM2A at band 5q31.3-33.1 (A pseudogene was mapped to chromosome 3.)
-
Gene product – GM2 activator
The gene is approximately 16 kb long.
The coding sequence encompasses 4 exons.
Table 4. Mutations in GM2A [14] (Open Table in a new window)
Mutation GM2A * |
Population |
Type |
E54X |
Laotian |
Infantile |
delK88, exon 3 |
Saudi-Arabian |
Infantile |
c.410delA, exon 3 |
Spanish |
Infantile |
C138R |
Black |
Infantile |
R169P, exon 4 |
Indian |
Infantile |
*All 5 known GM2 activator mutations are associated with acute GM2 gangliosidosis, each was found in a single patient. At least the first 4 were homozygotes, ie, they carried the same mutation on each of the 2 alleles. |
||
Workup
See workup and physical examination in the TSD section. Clinical DNA diagnostic evaluations for the GM2A mutation are now available (for more information, see Gene Tests).
Medical care
Medical care is as recommended for TSD.
Diagnostic Testing
At present, Clinical Laboratory Improvement Amendments (CLIA)–certified clinical diagnostic testing is widely available for TSD and SD (for more information, see Gene Tests). [15] CLIA-certified clinical diagnostic testing for hexosaminidase activator deficiency has also recently become available (for more information, see Gene Clinics).
Recent publications describe pharmacological chaperones that may stabilize and thereby increase the mutant ß-hexosaminidase A levels 3-fold to 6-fold in lysosomes of patients with TSD or SD. [16]
Whole exome sequencing (WES) is quickly becoming clinically available, and, with next-generation sequencing, the rate of throughput has increased exponentially. Previously, WES was avoided owing to its high cost and slow timescale in the return of results. Now that the cost of WES testing is substantially lower and it is quicker to return results, it has been adopted as a powerful tool in a diagnostician’s arsenal. [17, 18]
Retrospective immunohistochemical staining of brain specimens can be used to confirm diagnosis of GM2 gangliosidosis. [19]
Animal Models and Research
There is a major focus on animal research to elucidate many of the questions left unanswered involving the GM2 gangliosidoses. Several animal models have been developed, and investigations into the mechanisms involved, as well as therapeutic approaches, are underway in several laboratories internationally.
Mouse models of the GM2 gangliosidoses have been created. The mouse genome, about which much is known, is similar to the human genome.
Unlike human TSD and SD, the mouse models of TSD and SD exhibit very different neurologic phenotypes. While TSD mice usually have no neurologic abnormalities, the SD mice are very severely affected. Unlike human cells, mouse cells have 2 independent catabolic pathways to degrade GM2 gangliosides. In the pathway that is not present in human cells, GM2 is converted to GA2 by sialidase, and GA2 can be degraded by Hex B or Hex A. [20] Jacob sheep are the only animal model of TSD that exhibit clinical symptoms. [21]
TSD mice homozygous for defects in the HEXA gene (ie, knockouts) do accumulate GM2 ganglioside in MCBs, but regions of the nervous system show little storage because of Hex B involvement in the degradation pathway. However, pregnancy induces late-onset TSD because of reduced up-regulation of Hex B, which is a component of the bypass pathway. [22]
SD knockout mice are deficient in both Hex A and Hex B, and both degradation pathways are ineffective. The mice accumulate both GA2 and GM2, which leads to severe progressive neurologic defects by about age 3 months, imbalance and lack of coordination, and loss of limb movement. Significant accumulation of asialo-GM2 (GA2) occurs.
Double knockout mice have phenotypes, pathology, and biochemical features of the mucopolysaccharidoses. In TSD mice, Hex S provides enough activity to prevent a significant accumulation of glycosaminoglycan and a mucopolysaccharidosis phenotype.
SD mice treated with bone marrow transplantation (BMT) and by deprivation of a glycosphingolipid biosynthesis inhibitor survived significantly longer than those treated either with BMT or by substrate deprivation. In another experiment, intracerebral injection of low doses of adenovirus and the β -subunit of HEX B along with hyperosmotic concentrations of mannitol resulted in near-normal enzyme levels in the entire brain.
SD mouse modeling has yielded evidence for a two-stage process in disease progression. First, there is an activation of microgliosis, which is followed by later onset of astrogliosis, apoptosis, and a reduction in GluR1 AMPARs. This finding indicates an immunological role in SD and that excitotoxicity is not likely the cause of the neuronal death. [23]
Using a feline model of SD, researchers have been able to use adeno-associated viral (AAV) vectors to deliver feline HEX intracranially. This study showed that, following AAV treatment, microglial activation was repressed and neuroinflammatory marker levels were normalized, indicating that AAV gene delivery can mediate restoration of enzymatic activity and mitigate the significant inflammatory response in SD. [24]
Further research of SD mice may lead to new therapeutic approaches for GM2 gangliosidoses and to elucidation of the molecular mechanisms that cause neurodegeneration.
In addition to genetic modeling of GM2 gangliosidosis, the disease has been found in dogs and cats. In particular, there have been several cases of Shiba Inu dogs with deletions in the HEXB gene, resulting in Sandhoff disease phenotype. [25, 26]
Further Information
Carrier screening
The Committee on Genetics developed by the American College of Obstetricians and Gynecologists suggests that carrier screening be performed on any individual who may be at a heightened risk for a genetic condition such as TSD. In particular, couples who are of Ashkenazi Jewish, French-Canadian, or Cajun descent should be offered the option to undergo genetic screen when considering pregnancy or when pregnancy has already commenced. [27]
Support groups
Center for Jewish Genetic Diseases
Mount Sinai School of Medicine
Box 1497, One Gustave L. Levy Place
New York, NY 10029
Telephone: (212) 241-6947 (Consultation/Screening)
Center for the Study and Treatment of Jewish Genetic Diseases at the University of Pittsburgh Medical Center Health Systems
Telephone: (800) 334-7980
For information on testing, contact Erin O’Rourke, MS (eorourke@helix.hgen.pitt.edu)
CLIMB (Children Living with Inherited Metabolic Diseases)
Climb Building, 176 Nantwich Road
Crewe, CW2 6BG, United Kingdom
Telephone: 0845 241-2172 or 0845 241-2173 (Parents or Professionals)
Fax: 0845 241-2174
Email: fam.svcs@climb.org.uk (Family Services); cya.svcs@climb.org.uk (Children and Young Peoples Services); ir.svcs@climb.org.uk (Information Research)
Late-Onset Tay-Sachs Foundation
PO Box 5
Flourtown, PA 19031-0005
Telephone: (800) 672-2022 or (610) 292-9306
Email: lostf@verizon.net
1275 Mamaroneck Avenue
White Plains, NY 10605
Telephone: (914) 997-4488
Metabolic Information Network
PO Box 670847
Dallas, TX 75367-0847
Telephone: (214) 696-2188 or (800) 945-2188
Email: mizeg@ix.netcom.com
National Foundation for Jewish Genetic Diseases
250 Park Avenue, Suite 1000
New York, NY 10177
Telephone: (212) 371-1030
National Tay-Sachs and Allied Diseases Association (NTSAD)
2001 Beacon Street, Suite 204
Brighton, MA 02135
Telephone: (800) 906-8723
Fax: (617) 277-0134
Email: info@ntsad.org
1202 Lexington Avenue #288
New York, NY 10028
Telephone: (888) 354-7788 or (212) 431-0431
Fax: (888) 354-4884
NIH/National Institute of Neurological Disorders and Stroke
NIH Neurological Institute
PO Box 5801
Bethesda, MD 20824
Telephone: (800) 352-9424 or (301) 496-5751; TTY (301) 468-5981
Genetic information
Online Mendelian Inheritance in Man, OMIM
The Human Gene Mutation Database, HEXA
-
Fundus photograph showing retina changes associated with GM2 gangliosidosis.
Tables
Gene Features |
HEXA |
HEXB |
GM2A |
Chromosome location |
Band 15q23-q24 |
Band 5q13 |
Band 5q31.3-q33.1 |
Product |
Alpha subunit of Hex A; subunits of Hex S |
Beta subunit of Hex A; subunits of Hex B |
GM2 activator protein |
Heat sensitivity, pH |
Heat labile, acidic |
Heat stabile, basic |
Heat stabile, acidic |
TSD-B variant TSD-pseudo-AB variant classic infantile acute TSD |
Most severe phenotype; both alleles absent or mutated (deficient Hex A; may lead to increased levels of Hex B) |
Normal |
Normal |
B1 variant TSD |
Mutated (near-normal Hex A is inactive toward GM2) |
Normal |
Normal |
Adult chronic-type TSD |
Mutated, pseudodeficiency mutation in at least 1 allele |
Normal |
Normal |
SD, O variant |
Normal |
Both alleles absent or mutated (deficient Hex B; may lead to some Hex S activity) |
Normal |
Hexosaminidase Paris (SD) |
Normal |
At least 1 mutated allele (some normal Hex B activity) |
Normal |
AB variant (hexosaminidase activator deficiency) |
Normal (increased amounts of product) |
Normal (increased amounts of product) |
Absent or mutated |
Known mutations |
105 |
31 |
5 |
Group |
HEXA Mutation |
Frequency in Carriers |
Type |
Ashkenazi Jews |
1278+TATC, exon 11 |
75-80% |
Death in infancy |
|
IVS12+1G>C, intron 12 splice site mutation leads to a 35-bp deletion |
15-18% |
Death in infancy |
|
G269S, exon 7 |
3% |
Adult onset, variable phenotypes (homozygotes or compound heterozygotes, usually with 1278+TATC or IVS12+1G>S) |
|
R247W or R249W, exon 7 |
2% |
Benign pseudodeficiency |
Non-Ashkenazi Jews |
delF304, exon 8 |
Moroccan Jews, French, Italian, Portuguese (percentage not known) |
Death in infancy |
Non-Jewish enzyme- deficient TSD carriers |
1278+TATC, exon 11 |
5% in French Canadians and Acadians of Louisiana and 20% of non-Jewish European-derived populations |
Death in infancy |
|
5'UTRdel, 7.6-kb deletion (no mRNA) |
80% in non-Jewish French Canadians from eastern Quebec |
Death in infancy (first TSD mutation discovered) |
|
IVS9+1G>A, intron 9 splice site mutation |
15% in northern Europeans (French, Celtic), Cajun, and Pennsylvania Dutch |
Death in infancy |
|
IVS12+1G>C, intron 12 |
< 1% |
Death in infancy |
|
IVS5-1G>T |
Japanese most affected (no known percentage) |
Death in infancy |
|
12-bp del, exon 10 |
Turkish |
Death in infancy |
|
G454D, exon 12 |
Turkish |
Death in infancy |
|
R178H, exon 5 |
Northern Portuguese and unrelated Europeans in Mediterranean region |
B1 variant, severe late infantile (heterozygous with mRNA negative mutation or severe juvenile (homoallelic) |
|
R178L, exon 5 |
English |
Between infantile and B1 variant |
|
R178C, exon 5 |
Czechoslovakian |
B1 variant |
|
G250D, exon 7 |
Lebanese |
Juvenile |
|
D258H, exon 7 |
Scottish-Irish |
B1 variant |
|
R499H, exon 13 |
Mixed Jewish/Scottish-Irish |
Juvenile, early onset, rapidly progressive |
|
R504H, exon 13 |
Assyrian, Armenian, Lebanese, East European |
Juvenile |
|
G269S, exon 7 |
5% in Americans and Europeans |
Adult onset, variable phenotypes (homozygotes or compound heterozygotes with infantile TDS mutation) |
|
R247W or R249W, exon 7 |
35% |
Benign pseudodeficiency |
|
G250S |
80% in French Canadians |
Adult onset |
|
W474C |
|
Juvenile, later onset |
*More than 100 HEXA mutations spanning all 14 exons of the gene have been reported; approximately 70% cause acute TSD, approximately 20% cause subacute TSD, and approximately 9% cause chronic TSD. Approximately 58% of mutations are missense or nonsense mutations. Some genotype-phenotype correlations are described in Online Mendelian Inheritance in Man (OMIM). See also The Human Gene Mutation Database, HEXA. |
|||
HEXB Mutation |
Frequency in Carriers |
Type |
16-kb deletion (5' end to IVS5, reported as 50-kb deletion by some investigators) |
30% in French or French Canadians |
All types of SD |
50-kb deletion (5' end to IVS6) |
... |
Infantile |
Del76A frameshift mutation, exon 1 |
84% of subtype, Greek-Cypriot Maronite |
Infantile |
S62L, exon 1, and partial deletion compound |
... |
Infantile |
IVS2+1G>A |
Cordoba region of Argentina in mixed Creole, Spanish, and native peoples |
Infantile |
IVS8+5G>C splice site mutation |
Greek-Cypriot |
Infantile |
P417L, exon 11 |
Japanese, Italian, French Canadians |
Juvenile and some infantile (in compound with S255R) |
Y456S, exon 11 |
... |
Juvenile in compound heterozygote |
-26IVS12 24-bp insertion |
... |
Paris†, juvenile or asymptomatic, latter likely due to a second mutation |
-16IVS13 18-bp insertion, splice site mutation |
French |
Paris†, juvenile to asymptomatic |
P417L, exon 11, and 16/50-kb deletion compound |
Japanese, Italian, French Canadian |
Adult |
P504S, 16/50-kb deletion compound |
... |
Adult |
R505Q, exon 13, and 16/50-kb deletion compound (heat labile) |
... |
Adult |
R533H, exon 13 |
Japanese |
Adult (in compound with IVS2+1G>A) |
*Of 31 mutations, approximately 65% caused acute SD, approximately 17% caused subacute SD, approximately 13% caused chronic SD, and approximately 4% caused benign SD. Of the mutations, 42% were missense or nonsense mutations. † Hex A plus/Hex B minus was originally called Hexosaminidase Paris. See also Online Mendelian Inheritance in Man (OMIM) and The Human Gene Mutation Database, HEXB. |
||
Mutation GM2A * |
Population |
Type |
E54X |
Laotian |
Infantile |
delK88, exon 3 |
Saudi-Arabian |
Infantile |
c.410delA, exon 3 |
Spanish |
Infantile |
C138R |
Black |
Infantile |
R169P, exon 4 |
Indian |
Infantile |
*All 5 known GM2 activator mutations are associated with acute GM2 gangliosidosis, each was found in a single patient. At least the first 4 were homozygotes, ie, they carried the same mutation on each of the 2 alleles. |
||

What would you like to print?
- Introduction And Epidemiology
- Tay-Sachs Disease - GM2 Gangliosidosis Type I, Type III, Chronic, And B1 Variant
- Sandhoff Disease - GM2 Gangliosidosis Type II
- Hexosaminidase Activator Deficiency, GM2 Gangliosidosis, Type A B
- Diagnostic Testing
- Animal Models and Research
- Further Information
- Show All
- Tables
- References





