Background
Bruton agammaglobulinemia (see the image below) was the first primary immunodeficiency disease to be described. In 1952, Colonel Ogden Bruton noted the absence of immunoglobulins (Ig) in a boy with a history of pneumonia and other bacterial sinopulmonary infections. [1] Bruton was also the first physician to provide specific immunotherapy for this X-linked disorder by administering intramuscular injections of IgG. The patient improved but succumbed to chronic pulmonary disease in his fourth decade of life. Several historical reviews have been recently written. [2, 3]
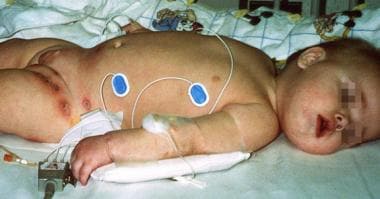 This patient presented with recurrent otitis and areas of cellulitis in the diaper area. Pseudomonas aeruginosa and Staphylococcus aureus were isolated from the skin lesions. Autoimmune hemolytic anemia and autoimmune neutropenia were confirmed based on the presence of autoantibodies. The patient has a mutation on exon 15, A504T, which changed an asparagine residue to a valine residue.
This patient presented with recurrent otitis and areas of cellulitis in the diaper area. Pseudomonas aeruginosa and Staphylococcus aureus were isolated from the skin lesions. Autoimmune hemolytic anemia and autoimmune neutropenia were confirmed based on the presence of autoantibodies. The patient has a mutation on exon 15, A504T, which changed an asparagine residue to a valine residue.
This disorder is now formally referred to as X-linked agammaglobulinemia (XLA), and the gene defect has been mapped to the gene that codes for Bruton tyrosine kinase (Btk) at band Xq21.3. The BTK gene is large and consists of 19 exons that encode the 659 amino acids that form the Btk cytosolic tyrosine kinase. Mutations can occur in any area of the gene. Btk is required for the proliferation and differentiation of B lymphocytes. [4, 5, 6]
In the absence of functional Btk, mature B cells that express surface Ig and the marker CD19 are few to absent. The absence of CD19 is readily detected with fluorocytometric assays, and this finding usually easily confirms the diagnosis of XLA in a male. As Bruton originally described, XLA manifests as pneumonia and other bacterial sinopulmonary infections in 80% of cases. [1] Such infections that begin in male infants as maternal IgG antibodies, acquired transplacentally, are lost. Thus, XLA is most likely to be diagnosed when unusually severe or recurrent sinopulmonary infections occur in a male infant younger than 1 year.
In some individuals, the diagnosis is delayed into adulthood. In some cases, this delay can be explained by the variable severity of XLA, even within families in which the same mutation is present. However, a significant contributing factor is the deceptively poor inflammatory response seen in the absence of antibodies. Delayed diagnosis puts patients at risk for chronic pulmonary disease and poor growth, leading to mortality at a younger age. Encapsulated bacteria, most commonly Streptococcus pneumoniae, followed by Haemophilus influenzae type b and staphylococcal species, are the typical pathogens.
Pathophysiology
In the absence of mature B cells, patients lack lymphoid tissue and fail to develop plasma cells, the cells that manufacture antibodies. Germinal centers where B cells proliferate and differentiate are poorly developed in all lymphoid tissue, including the spleen. Tonsils, adenoids, peripheral lymph nodes, and Peyer patches in the intestines are all small or absent. The lungs and the lamina propria of the gut lack the normal pattern of lymphocyte distribution. However, biopsy of lymphoid tissue and bone marrow examination are not currently performed in the workup of most cases of XLA.
Animal models of human BTK mutations are confined to mice at this time. Mouse models have milder disease than humans. However, murine models, including knockout and transgenic mice, have been useful in understanding the mechanisms of B lymphopoiesis, B-cell differentiation, and antibody formation. Murine gene mutations in human counterparts may be associated with a clinical illness different from the illness seen in mice.
Bruton's tyrosine kinase (Btk) is a member of the Tec family of kinases. Activation of Btk results in a cascade of signaling events resulting in calcium mobilization and fluxes, cytoskeletal rearrangements, and transcriptional regulation involving nuclear factor-kappaB (NF-kappaB) and nuclear factor of activated T cells (NFAT). Its activation is tightly controlled by numerous other signaling proteins including protein kinase C (PKC), Sab/SH3BP, and caveolin-1. [7]
Although defects may occur in many steps in B-cell development and maturation, resulting in agammaglobulinemia, the most common and well-described defect is the impaired maturation of the pro–B cells to pre–B cells. In the fetal bone marrow, the first committed cell in B-cell lineage is the early pro–B cell, which is identified by its ability to proliferate in the presence of interleukin (IL)-7. These cells develop into late pro–B cells, in which rearrangement of the heavy chain occurs. This rearrangement process requires the recombination-activating genes (ie, RAG1 and RAG2); their enzymatic activities are controlled by IL-7 and, perhaps, by other factors.
When the heavy chain is produced, it is transported to the cell surface by IgA (CD79a) and IgB (CD82) heterodimers or by the surrogate light chain. Progression from this late pro–B-cell stage to the pre–B-cell stage involves the rearrangement and joining of the various segments of the heavy chain. The completion of light- and heavy-chain rearrangement and the presence of surface IgM results in an immature B cell, which then leaves the bone marrow.
Increasing levels of IgD in the transitional cells finally results in the mature B cell, with both IgM and IgD expressed. The mature B cells circulate between secondary lymphoid organs and migrate into lymphoid follicles of the spleen and lymph nodes in response to further stimuli and various chemokines. T cells stimulate B cells to undergo further proliferation and Ig class switching, leading to the expression of the various isotypes of Ig (ie, IgG, IgA, or IgE). Activation of the B-cell receptor (BCR) induces the recruitment of Syk, which phosphorylates BLNK, a contributor to the activation of BTK that affects other intracellular signaling events.
Murine B-cell proliferation and differentiation is under the control of BTK, as well as SYK; PAX5; and genes that code for l5, Ig-a, Ig-b, g chain of IL-2 receptor (IL-2Rg), lyn, and bcl-2. Mutations in these mouse genes and in the mouse gene for Btk lead to milder forms of B-cell deficiency compared with that of humans with BTK, m heavy-chain (µH), or l5 mutations.
Mutations in the murine IL receptor common g chain also cause mild B-cell deficiency in mice. In contrast, mutations in the human IL common g chain cause X-linked severe combined immunodeficiency (SCID), with normal-to-high levels of B cells expressing CD19. These findings indicate that a defect in any of the steps in B-cell development may be clinically important. Approximately 85% of patients with defects in early B-cell development have XLA.
The vital role of Btk in Toll-like receptor (TLR) signaling has been supported by several lines of evidence. Although patients with XLA have normal numbers of circulating dendritic cells, a profound impairment of IL-6 and tumor necrosis factor (TNF)-a production is observed in response to the TLR8 agonist ssRNA. [8, 9] This may provide an explanation for their increased susceptibility to enteroviral infections. Others have also found defective TLR2, TLR4, and TLR7/8–induced TNF-a production. [10] Btk is involved in TLR9 activation and expression [11] and TLR-induced IL-10 production. [12] Subsequent involvement of other immune cells such as NK may be effected by Btk regulation of IL-12 and IL-18 production. [13] It may be that Btk is required for hem oxygenase-1 gene activation by major TLR pathways. [14]
On the other hand, mutations in the Btk gene do not always result in X-linked agammaglobulinemia, and measurements of actual Btk protein blood levels and/or measurements of B-cell numbers, as well as more advanced genetic assays, may be needed to confirm the diagnosis. [15]
More than 600 patients with mutations in Btk involving all functional domains have been reported, and additional new disease-causing variants continue to be discovered. Insertions and deletions of less than 5 bp and single-base-pair substitutions are responsible for more than 90% of documented mutations.
Epidemiology
Old content
Frequency
United States
The estimated birth rate of XLA in the United States was calculated to be 1 case per 379,000 live births. A prevalence of 1 case per 250,000 individuals has been estimated in the United States. However, this number was reviewed prior to the availability of mutational analysis and is generally considered to be an underestimate. New mutations are believed to cause 30-50% of XLA cases.
International
Geneticists believe that the prevalence of XLA is similar among most ethnic groups. The prevalence of XLA in Eastern and Central Europe is 1 per 1,400,000. [16] Data from France have suggested a prevalence of 1 case per 70,000-90,000 population. The greater frequency in France may well be related to more accurate acquisition of statistics. Black, Japanese, and Malaysian populations have lower reported frequencies of clinical XLA, but whether these frequencies are accurate is debatable because of the genetic mechanisms that cause XLA.
The specific mutations of Btk have been examined in Mexican, [17] Brazilian, [18] Iranian, [13] and Chinese [19] populations. The experience of a single center in India has been published. [20]
Mortality/Morbidity
Patients who received intravenous IgG (IVIG) before age 5 years have lower morbidity and mortality rates than previously identified patients who were treated only with fresh-frozen plasma (FFP) and intramuscular Ig (IMIG); achieving IgG levels near normal or even above 200 mg/dL is difficult using FFP or IMIG. Patients who receive IVIG or subcutaneous IgG (SCIG) therapy regularly may have a near-normal lifestyle. Patients are known to survive into their seventy's year of life.
Viral and pulmonary infections cause more than 90% of mortalities.
Chronic enteroviral infections are the most common etiology for early morbidity. High-dose IVIG or Ig administered intrathecally has slowed, but not stopped, the progression of CNS deterioration. Dementia, ataxia, and paresthesias are the common clinical features of meningoencephalitis due to enteroviruses. Other viral causes of death are sporadic. Adenoviruses are well-recognized causes of morbidity and mortality in any patient with immunocompromise. Hepatitis viruses are also a risk; hepatitis C has been transmitted by IVIG preparations with inadequate viral inactivation processes. Overall, viral infections resulted in one half of the deaths that occurred in 3 series.
Pulmonary infections, both acute and chronic, account for most other deaths. Recurrent pulmonary infections frequently lead to bronchiectasis. Common causative agents include S pneumoniae, H influenzae type b, and Staphylococcus aureus. Burkholderia cepacia and coagulase-negative staphylococci are other significant bacterial agents.
If present, inflammatory bowel disease is usually chronic in XLA and leads to malnutrition and cachexia and further increases the risk of infection. Other autoimmune disorders such as arthritis can be especially disabling. In patients with these complications, examining for involvement of microbial pathogens is important because treatment with immunosuppressive medications may complicate their management.
Race
Most investigators have studied northern European populations. Although black, Japanese, and Malaysian populations are reported to have lower risks for XLA, geneticists doubt the accuracy of these statistics. Recently, more reports have detailed Asian [21, 22] and Arabic populations. [23]
Sex
XLA is a disorder that affects only males. No carrier female with any clinical illness related to the mutated allele has been identified. Girls with absent mature B cells may have autosomal recessive mutations that affect gene products other than those of BTK (see Agammaglobulinemia).
Age
Because XLA is a genetic disorder, male infants can be identified with prenatal diagnosis when the mother has been identified as a carrier. Chorionic villus sampling (CVS) can be performed early in pregnancy, and DNA analysis can be used when the family's exact mutation is known. Amniocentesis can be performed later in gestation. Collection of fetal lymphocytes through in utero umbilical cord sampling can be used to enumerate CD19+ B cells and mature T cells using fluorocytometric analysis, although this procedure places the fetus at some risk for mortality (ranging from < 1-5%). At birth, cord blood can be sent for fluorocytometric analysis of lymphocyte populations. Quantitative IgG levels are not useful; cord and fetal IgG levels largely reflect maternal IgG transported across the placenta.
Because of passive transplacental acquisition of maternal IgG, newborns have normal serum IgG levels and may not have problems until the IgG is catabolized. Because newborns cannot produce their own Ig, increased susceptibility to infections usually develops in infants older than about 6 months. Therefore, patients with XLA can clinically present when they are aged 3 months to 5 years. Most cases of XLA are now identified in patients younger than 1 year, depending on the rate of maternal-derived IgG loss and occurrence of infections. The average age of diagnosis is younger in patients with a family history (2.6 y) than in those without (5.4 y).
Patients may also present in the second or third decade of life, although this is uncommon. The oldest age at diagnosis was 51 years. These patients may have milder disease related to the presence of mutated Btk protein rather than complete absence of the protein. Rarely, the individual has mild disease while others with the same mutation have more severe clinical illness.
New content
Primary Agammaglobulinemia is a rare disorder that occurs almost exclusively in males although some females have been affected by certain types of this disorder. The incidence of XLA in the United States has been estimated to be at least 1/190,000 male births. The age of onset of symptoms for most patients is between 3 months and 3 years, with over 50% of patients becoming symptomatic by 1 year of age and more than 90% of patients becoming symptomatic by 5 years of age. [24, 25]
-
This patient presented with recurrent otitis and areas of cellulitis in the diaper area. Pseudomonas aeruginosa and Staphylococcus aureus were isolated from the skin lesions. Autoimmune hemolytic anemia and autoimmune neutropenia were confirmed based on the presence of autoantibodies. The patient has a mutation on exon 15, A504T, which changed an asparagine residue to a valine residue.
-
Bruton agammaglobulinemia (ie, X-linked agammaglobulinemia [XLA]) in brothers. XLA was diagnosed in the less-robust younger brother when he presented with neutropenia and typhlitis. The older brother, with a history of 7 episodes of pneumonia, was then evaluated and diagnosed with XLA. In both brothers CD19- B cells were less than 1%; this finding is consistent with XLA.
Tables
Brand(Manufacturer) |
Manufacturing Process |
pH |
Additives (IVIG products containing sucrose are more often associated with renal dysfunction, acute renal failure, and osmotic nephrosis, particularly with preexisting risk factors [eg, history of renal insufficiency, diabetes mellitus, age >65 y, dehydration, sepsis, paraproteinemia, nephrotoxic drugs].) |
Parenteral Form and Final Concentrations |
IgA Content mcg/mL |
Carimune NF (ZLB Behring) |
Kistler-Nitschmann fractionation, pH 4 incubation, nanofiltration |
6.4-6.8 |
6% solution: 10% sucrose, < 20 mg NaCl/g protein |
Lyophilized powder 3, 6, 9, 12% |
Trace |
Flebogamma (Grifols USA) |
Cohn-Oncley fractionation, PEG precipitation, ion-exchange chromatography, pasteurization |
5.1-6 |
Sucrose free, contains 5% D-sorbitol |
Liquid 5% |
< 50 |
Gammagard Liquid 10% (Baxter Bioscience) |
Cohn-Oncley cold ethanol fractionation, cation and anion exchange chromatography, solvent detergent treated, nanofiltration, low pH incubation |
4.6-5.1 |
0.25 M glycine |
Ready-for-use liquid 10% |
37 |
Gammar-P IV (ZLB Behring) |
Cohn-Oncley fraction II/III, ultrafiltration, pasteurization |
6.4-7.2 |
5% solution: 5% sucrose, 3% albumin, 0.5% NaCl |
Lyophilized powder 5% |
< 20 |
Gamunex (Talecris Biotherapeutics) |
Cohn-Oncley fractionation, caprylate-chromatography purification, cloth and depth filtration, low pH incubation |
4-4.5 |
Contains no sugar, contains glycine |
Liquid 10% |
46 |
Gammaplex (Bio Products) |
Solvent/detergent treatment targeted to enveloped viruses; virus filtration using Pall Ultipor to remove small viruses including nonenveloped viruses; low pH incubation |
4.8-5.1 |
Contains sorbitol (40 mg/mL); do not administer if fructose intolerant |
Ready-for-use solution 5% |
< 10 |
Iveegam EN (Baxter Bioscience) |
Cohn-Oncley fraction II/III, ultrafiltration, pasteurization |
6.4-7.2 |
5% solution: 5% glucose, 0.3% NaCl |
Lyophilized powder 5% |
< 10 |
Polygam S/D Gammagard S/D (Baxter Bioscience for the American Red Cross) |
Cohn-Oncley cold ethanol fractionation followed by ultracentrafiltration and ion exchange chromatography, solvent detergent treated |
6.4-7.2 |
5% solution: 0.3% albumin, 2.25% glycine, 2% glucose |
Lyophilized powder 5%, 10% |
< 1.6 (5% solution) |
Octagam (Octapharma USA) 9/24/10: Withdrawn from market because of unexplained reports of thromboembolic events |
Cohn-Oncley fraction II/III, ultrafiltration, low pH incubation, S/D treatment pasteurization |
5.1-6 |
10% maltose |
Liquid 5% |
200 |
Panglobulin (Swiss Red Cross for the American Red Cross) |
Kistler-Nitschmann fractionation, pH 4 incubation, trace pepsin, nanofiltration |
6.6 |
Per gram of IgG: 1.67 g sucrose,< 20 mg NaCl |
Lyophilized powder 3, 6, 9, 12% |
720 |
Privigen (CSL Behring) |
pH 4 incubation, octanoic acid fractionation, depth filtration, and virus filtration |
4.6-5 |
10% solution; Preservative-free and sucrose- and maltose-free |
Ready-to-use solution 10% |
25 |
Brand(Manufacturer) |
Manufacturing Process |
pH |
Additives |
Parenteral Form and Final Concentrations |
IgA Content mcg/mL |
Vivaglobin (ZLB Behring) |
Cold ethanol fractionation, pasteurization |
6.4-7.2 |
2.25% glycine, 0.3% NaCl |
Liquid 16% (160 mg/mL) |
< 50 mcg/mL |




