Practice Essentials
Ornithine transcarbamylase (OTC) deficiency is an X-linked genetic disorder of the urea cycle that leads to elevated levels of ammonia. It can present as a severe neonatal-onset disease (primarily in males) or late onset disease in males and females. [1]
See the image below.
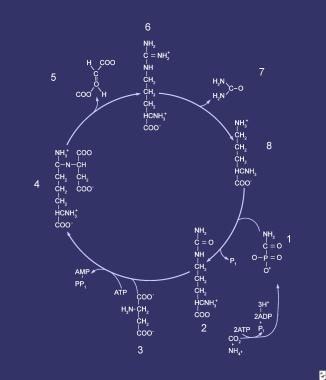 Compounds that comprise the urea cycle are sequentially numbered, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; during this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamoyl phosphate synthetase (CPS). Compound 2 is citrulline, which is the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Compounds that comprise the urea cycle are sequentially numbered, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; during this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamoyl phosphate synthetase (CPS). Compound 2 is citrulline, which is the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Signs and symptoms
Hyperammonemia from OTC deficiency can be manifested clinically by some or all of the following:
-
Anorexia
-
Irritability
-
Heavy or rapid breathing
-
Lethargy
-
Vomiting
-
Disorientation
-
Somnolence
-
Asterixis (rare)
-
Combativeness
-
Obtundation
-
Coma [2]
-
Cerebral edema
-
Death (if treatment is not forthcoming or effective)
Presentation in patients with severe neonatal-onset phenotype may be as follows:
-
Severely affected infants (typically male hemizygotes) will be well appearing at birth
-
Reduced feeding and emesis in the first week of life (typically Day 2 or 3), followed by encephalopathy due to hyperammonemic crisis
-
Hyperammonemic infants are at risk for subclinical seizures
Presentation in patients with late-onset phenotypes (males or females):
-
Intermittent encephalopathy or psychotic symptoms in particular after metabolic stress, such as an intercurrent illness, accident or drastic dietary changes (fasting or excessive protein intake)
-
Recurrent vomiting
-
Anorexia, protein aversion or avoidance
-
Migraine headaches, in particular in heterozygous females after excessive protein intake [3]
-
Seizures
Physical findings may include the following:
-
Poor growth
-
Tachypnea or hyperpnea while hyperammonemic
-
Apnea and respiratory failure, in the latter stages of disease progression
Neurologic findings include the following:
-
Poor coordination
-
Dysdiadochokinesia
-
Hypotonia or hypertonia
-
Ataxia
-
Tremor
-
Lethargy that may progress to combativeness, obtundation, and coma if hyperammonemia worsens
-
Decorticate or decerebrate posturing in cases of severe hyperammonemia
See Clinical Presentation for more detail.
Diagnosis
In patients with OTC deficiency, molecular testing identifies a pathogenic variant in the OTC gene in 80-90% of cases. Sequence analysis is diagnostic in 80% of probands, and deletion/duplication analysis identifies the etiology in an additional 5-10% of cases. [4]
Other laboratory findings may include the following:
-
Markedly elevated ammonia levels. During a hyperammonemic crisis, ammonia levels may exceed 2,000 mg/dL
-
Plasma amino acid analysis with elevated glutamine (>800 µmol/L) and low citrulline (< 10 nmol/mL)
-
Elevated urinary orotic acid level
-
Low blood urea nitrogen (BUN) level
-
Normal liver and kidney function in most cases, unless hypoxia or shock supervene
-
Reduced OTC enzymatic activity in biopsied liver tissue. Symptomatic patients typically have enzymatic activity less than 30% of control, with severe infantile cases having activity less than 20% control. Note that liver biopsy is not recommended in female patients due to random X-inactivation posing risk for false negative results.
See Workup for more detail.
Management
Treatment of OTC deficiency during metabolic decompensation consists of the following:
-
Immediate temporary discontinuation of protein intake
-
Compensatory increases in dietary carbohydrates and lipids
-
Intravenous administration of sodium benzoate, sodium phenylacetate and arginine
-
Hemodialysis as needed for rapid correction of severe hyperammonemia
Longterm treatment of OTC deficiency consists of the following:
-
Dietary protein restriction based on individual protein tolerance
-
Ammonia scavenging medications and citrulline or arginine supplementation as required
-
Liver transplantation in severe neonatal cases or cases refractory to medical management
Agents to avoid include the following:
-
Valproate
-
Haloperidol
-
Systemic corticosteroids
See Treatment and Medication for more detail.
Background
Ornithine transcarbamylase (OTC) deficiency is the most common urea cycle disorder. The OTC enzyme is responsible for condensation of carbamyl phosphate and ornithine to form citrulline. When OTC enzymatic activity is reduced, this leads to dimished ammonia incorporation and therefore, hyperammonemia (see Hyperammonemia). OTC is hepatically expressed and is intramitochondrial. [5] The OTC gene is X-linked.To date, more than 400 disease-causing mutations have been reported. [4]
Pathophysiology
The hepatic urea cycle is the major route for waste nitrogen disposal, which is chiefly generated by protein and amino acid metabolism. [6] Low-level synthesis of certain cycle intermediates in extrahepatic tissues makes a small contribution to waste nitrogen disposal. A portion of the cycle is mitochondrial in nature; mitochondrial dysfunction may impair urea production and result in hyperammonemia. [7] Overall, activity of the cycle is regulated by the rate of synthesis of N -acetylglutamate, the enzyme activator that initiates incorporation of ammonia into the cycle.
Failure to incorporate carbamyl phosphate into citrulline by condensation with ornithine results in an excess of both substrates for the reaction (see the image below).
Compounds that comprise the urea cycle are sequentially numbered, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; during this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamoyl phosphate synthetase (CPS). Compound 2 is citrulline, which is the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
The consequent increase in hepatic ornithine often is reflected in an elevated serum level. By contrast, excessive mitochondrial carbamoyl phosphate finds its way into the cytosol, where it functions as substrate for the carbamoyl phosphate synthetase (CPS) II reaction. This results in orotic acid, which is a normal intermediate in pyrimidine biosynthesis. Pyrimidine biosynthesis is regulated very tightly because it is a pathway involved in nucleic acid biosynthesis; thus, increases in urinary excretion of orotic acid rarely are observed in healthy humans.
Frequency
United States
OTC deficiency is the most common urea cycle disorder and may present after childhood in preivously healthy individuals. There is an estimated incidence rate of 1:56,500. [8] However, this may be an underestimation due to missed late-onset cases.
Mortality/Morbidity
Morbidity and mortality are high, especially in patients with the neonatal form. Close followup with a metabolic center is essential to optimizing outcomes. As an X-linked trait, severity of presentation in heterozygous females is determined in part by skewed X-chromosome inactivation. Females may range from asymptomatic to severely affected with risk for life-threatening hyperammonemia.
Prognosis
The long-term outlook for individuals with all forms of OTC deficiency is guarded, particularly with reference to cerebral function. [9] The prognosis for neonates with hyperammonemic coma is determined by the duration of hyperammonemia, rather than the peak ammonia level. [10]
Many heterozygous females appear to be relatively healthy, but on neuropsychologic testing are noted to have deficits in executive function and fine motor tasks. [11]
Patient Education
Family pedigree studies in this disease are essential for the following two reasons:
-
The X-linked nature of the mutation leads to a 1:2 chance of recurrence in any subsequent male conceptus if the mother is a heterozygote.
-
For inherited (non-spontaneous) disease, all female siblings of the mother are potential heterozygotes and male siblings may be at risk for late-onset presentation.
-
Compounds that comprise the urea cycle are sequentially numbered, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; during this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamoyl phosphate synthetase (CPS). Compound 2 is citrulline, which is the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.



