Background
Tetralogy of Fallot (TOF) with absent pulmonary valve is a rare congenital anomaly characterized by features of tetralogy of Fallot with either rudimentary ridges or the complete absence of pulmonic valve tissue and usually with a hypoplastic pulmonary valve annulus. Congenital absence of the pulmonary valve with an intact ventricular septum occurs, but this is much less common.
Since the absent pulmonary valve is the dominant feature, some authors have called this the absent pulmonary valve syndrome (APVS). APVS has been described to have three subtypes; two of these are very rare and include APVS with an intact septum and APVS with tricuspid atresia. APVS with tetralogy of Fallot is the most common subtype, accounting for over 80% of the cases. [1]
The absence of mature pulmonary valve tissue leads to severe pulmonary regurgitation, which is often associated with massive dilatation of the proximal branch pulmonary arteries and which is characteristic of this syndrome.
In a recent meta-analysis of 140 patients who underwent invasive testing, an abnormal karyotype was seen in 39%. [2] Another multicenter trial found that genetic abnormalities were seen in 46% of 80 patients tested, of which 22q11 deletion accounted for 35%. [3]
An interesting feature of this anomaly is that the ductus arteriosus is frequently absent. However, when the pulmonary valve is absent and the ventricular septum is intact, a normal ductus arteriosus is also generally present. [4]
The image below compares the pulmonary artery branching between healthy patients and those with absent pulmonary valve syndrome.
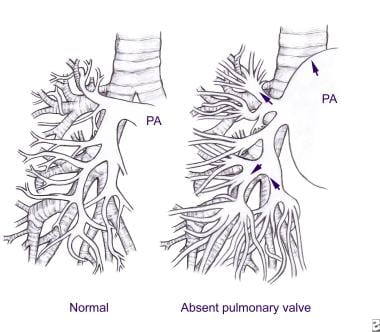 Tetralogy of Fallot With Absent Pulmonary Valve. Pulmonary artery branching in a healthy person and in a patient with absent pulmonary valve syndrome.
Tetralogy of Fallot With Absent Pulmonary Valve. Pulmonary artery branching in a healthy person and in a patient with absent pulmonary valve syndrome.
Tetralogy of Fallot is the most common cause of cyanotic heart disease and may occur at a rate of 1-3 cases per 1000 live births. However, tetralogy of Fallot with absent pulmonary valve is rare, with approximately 3% of patients with tetralogy of Fallot having the absent pulmonary valve syndrome.
See Tetralogy of Fallot With Pulmonary Stenosis, Tetralogy of Fallot With Pulmonary Atresia, and Tetralogy of Fallot in Adults for more information on these topics.
Pathophysiology
Tetralogy of Fallot (TOF) consists of a malalignment ventricular septal defect (VSD), infundibular pulmonary stenosis, overriding aorta, and right ventricular hypertrophy (RVH).
The absence of a functioning pulmonary valve gives rise to pulmonary regurgitation (insufficiency) that results in aneurysmal dilatation of the main and branch pulmonary arteries, which can compress the tracheobronchial tree (see the following image). In addition to compression of the larger bronchi, Rabinovitch et al described abnormal tufts of the smaller pulmonary arteries that compress the intrapulmonary bronchi. [5] The investigators further reported a reduction of the number of alveoli. This may explain why surgical relief of the larger airway compression alone is not always effective in reversing the severe obstructive respiratory disease.
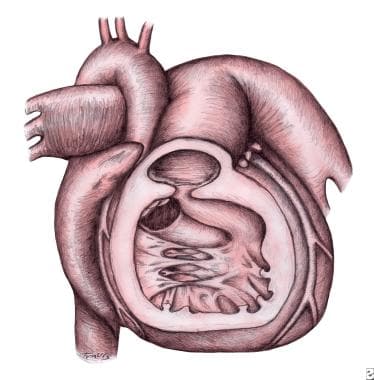 Tetralogy of Fallot With Absent Pulmonary Valve. Drawing showing absence of the pulmonary valve with features of tetralogy of Fallot. Note the small nubbins of tissue at the pulmonary valve annulus in the center of the drawing. Characteristic muscular right ventricular hypertrophy and infundibular pulmonary stenosis are present. A right aortic arch, a ventricular septal defect with overriding aortic valve, and massively dilated main and branch pulmonary arteries are present.
Tetralogy of Fallot With Absent Pulmonary Valve. Drawing showing absence of the pulmonary valve with features of tetralogy of Fallot. Note the small nubbins of tissue at the pulmonary valve annulus in the center of the drawing. Characteristic muscular right ventricular hypertrophy and infundibular pulmonary stenosis are present. A right aortic arch, a ventricular septal defect with overriding aortic valve, and massively dilated main and branch pulmonary arteries are present.
The pulmonary valve annulus is usually hypoplastic, resulting in pulmonary stenosis. The stenosis is not typically severe and the pathophysiology in this condition is such that, after the immediate neonatal period, a net left-to-right shunt is observed. This and the airway obstruction due to the dilated pulmonary arteries are the hallmarks of the condition.
In the immediate neonatal period, cyanosis may be present, which is a result of increased pulmonary vascular resistance (PVR) causing a right-to-left shunt at the level of the VSD or the inability to effectively ventilate the patient. After the fall in pulmonary vascular resistance, respiratory difficulties are the most prominent symptom in severe cases.
Etiology
Etiologic factors for tetralogy of Fallot (TOF) with absent pulmonary valve are not known in most cases. Chromosomal abnormalities, absence of the ductus arteriosus, and other theories have been proposed.
Chromosomal abnormalities
Absent pulmonary valve syndrome has been reported in association with chromosomal abnormalities that involve chromosomes 6 and 7, [6, 7] as well as in association with a deletion of chromosome 22 and DiGeorge syndrome in about 25% of cases. [8, 9, 10, 11] Two large studies have described genetic abnormalities in 40-46% of cases, with 22q11 deletion syndrome being the most common and noted in 35% of cases. [2, 3]
Absence of ductus arteriosus
Emmanoulides et al were the first to highlight the association of absent pulmonary valve syndrome with the absence of the ductus arteriosus. [12] The investigators proposed a pathogenetic link between the lack of the ductus arteriosus and pulmonary artery dilatation and the absent pulmonary valve. They argued that, because most of the blood that enters the pulmonary artery does not have the usual egress through the ductus arteriosus, the blood returns to the right ventricle (RV) through the somewhat stenotic pulmonic annulus; thus, it contributes to the dilatation of the pulmonary arteries and the possible nondevelopment of the pulmonic valve. [12] This blood then crosses the ventricular septal defect (VSD) to feed the lower resistance placental circulation through the left ventricle (LV).
However, this theory has been challenged on the basis that tetralogy of Fallot with absent pulmonary valve syndrome occasionally presents with a ductus arteriosus. [13] Ettedgui et al suggested poststenotic dilation as the mechanism for the dilated pulmonary arteries. [13]
One study reported that reversal of end-diastolic umbilical flow in fetuses at 10-14 weeks' gestation is a poor prognostic sign seen in patients with absent pulmonary valve and a patent ductus arteriosus; this suggests that the frequent absence of a ductus in later pregnancy in patients with tetralogy of Fallot with absent pulmonary valve is a result of preselection and that only those fetuses with a small or absent ductus arteriosus are able to survive to term. [14]
Other theories
Others have postulated that agenesis of the ductus arteriosus results from obliteration of a still immature artery slightly later in development rather than from complete failure of a sixth arch artery to develop. [15] Rabinovitch et al have suggested that some congenital weakness of the pulmonary arteries may be present, although the histologic findings are not specific. [5] Some authors believe that the described changes are the result of increased wall stress similar to the changes seen in pulmonary hypertensive arteriopathy; however, no such wall abnormalities have been demonstrated in peripheral pulmonary arteries.
In fetuses with a ductus arteriosus, the direction of flow through the ductus is controversial. Lakier et al concluded that the flow was from the pulmonary artery to the descending aorta because of lower placental resistance. [16] However, other authors dispute this and contend that the flow of blood occurs from the aorta into the pulmonary artery, and, because no pulmonic valve is present, the diastolic pressures between the aorta and the ventricles equalize, leading to ventricular dysfunction and impairment of the diastolic filling of the ventricles. [4, 17]
If the ventricular septum is intact, this affects only the RV and may be the pathogenetic mechanism of the membranous tricuspid atresia described in association with absent pulmonary valve and intact ventricular septum. [4, 18] Yeager et al suggest that tetralogy of Fallot with dysplastic pulmonary valve may be more common than apparent from clinical experience but that it is generally lethal in a fetus with a large ductus arteriosus, because the function of both ventricles is adversely affected. [4] Only fetuses in which the ductus is restrictive or absent are able to survive to term. [4]
Prognosis
Prognosis in patients with tetralogy of Fallot (TOF) with absent pulmonary valve is related to both the associated genetic abnormality and the degree of tracheobronchial obstruction secondary to pulmonary artery dilatation. Airway compromise is the predominant concern, including atelectasis, pneumothorax, and pneumonia.
Neonates with severe respiratory distress soon after birth are most at risk for early death. Infants who require surgery early in life have a worse prognosis than those repaired at a later date. [19, 20]
Galindo et al reported that many fetuses with absent pulmonary valve syndrome have an increased nuchal thickness in the first trimester and that the 22q11 microdeletion is the most common associated karyotype anomaly (21% of their patients); these findings suggest an extremely poor outlook in these patients (only 2 of 14 patients in the study ultimately survived). [8] Other authors have reported similar prognosis findings. [9, 21]
In a Japanese study, patent ductus arteriosus appeared to be a good prognostic factor for fetal and postnatal outcomes, whereas polyhydramnios, hydrops fetalis, and balloon type (vs clover type) pulmonary configuration were associated with poor prognosis. [22]
Overall, the prognosis remains poor with a high perinatal and neonatal mortality. Overall survival was reported at 26% [2] and 64% [3] in two large studies.
Morbidity/mortality
Mortality and morbidity rates in patients with tetralogy of Fallot with absent pulmonary valve syndrome far exceed those of patients with normal physiology who have typical tetralogy of Fallot. Patients are at risk for hypoxemia, heart failure, respiratory failure, and combinations of these events.
The size of the pulmonary valve annulus and, therefore, severity of pulmonary regurgitation substantially influence patient morbidity and mortality. Patients with a smaller, more stenotic annulus are subject to risks akin to those of typical tetralogy of Fallot. Patients with a large annulus and, therefore, more severe pulmonary regurgitation are at greater risk of morbidity and mortality. Patients with severe bronchial obstruction develop symptoms in the early neonatal period. As the airways increase in size and strength, these symptoms may decrease. However, this usually cannot be expected to occur until approximately age 9 months.
Hypoxemia
The newborn may demonstrate significant cyanosis until pulmonary vascular resistance (PVR) falls, after which the degree of hypoxemia reflects the severity of pulmonary annular stenosis. A larger pulmonary annulus produces less stenosis, and, therefore, intracardiac shunting may primarily be left-to-right, resulting in minimal cyanosis. The patient with more severe annular hypoplasia presents more similarly to the patient with typical tetralogy of Fallot.
Heart failure
Congestive heart failure (CHF) can occur as a result of a large left-to-right ventricular shunt. This contributes to an enlarged left atrium, which, along with dilated pulmonary arteries, results in airway compression. The presence of significant tricuspid regurgitation also increases the risk of heart failure.
Respiratory failure
In patients with more severe pulmonary regurgitation, aneurysmal dilation of pulmonary arteries can cause air trapping due to bronchial compression. This process can be localized or diffuse and may be severe.
-
Tetralogy of Fallot With Absent Pulmonary Valve. Drawing showing absence of the pulmonary valve with features of tetralogy of Fallot. Note the small nubbins of tissue at the pulmonary valve annulus in the center of the drawing. Characteristic muscular right ventricular hypertrophy and infundibular pulmonary stenosis are present. A right aortic arch, a ventricular septal defect with overriding aortic valve, and massively dilated main and branch pulmonary arteries are present.
-
Tetralogy of Fallot With Absent Pulmonary Valve. Pulmonary artery branching in a healthy person and in a patient with absent pulmonary valve syndrome.





