Practice Essentials
Wiskott-Aldrich syndrome (see the image below) is an X-linked recessive immunodeficiency disorder characterized in one third of patients by the triad of recurrent bacterial sinopulmonary infections, eczema (atopiclike dermatitis), and a bleeding diathesis caused by thrombocytopenia and platelet dysfunction. [1]
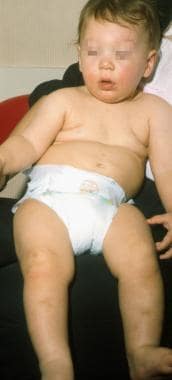 This 10-month-old infant presented with bloody diarrhea at age 4 months followed by recurrent otitis media infections. A maternal uncle had Wiskott-Aldrich Syndrome (WAS). Note the mild malar eczema and pretibial ecchymoses in this nonambulatory child. His diagnosis was confirmed by immunologic parameters, thrombocytopenia, and low platelet volume.
This 10-month-old infant presented with bloody diarrhea at age 4 months followed by recurrent otitis media infections. A maternal uncle had Wiskott-Aldrich Syndrome (WAS). Note the mild malar eczema and pretibial ecchymoses in this nonambulatory child. His diagnosis was confirmed by immunologic parameters, thrombocytopenia, and low platelet volume.
Signs and symptoms
The characteristic triad of bleeding, eczema, and recurrent infections in Wiskott-Aldrich syndrome generally become evident during the first year of life, with petechiae and ecchymoses of the skin and oral mucosa and bloody diarrhea being the first clinical signs. Although only one third of patients with WASP (Wiskott-Aldrich syndrome protein) mutations express the classic triad at presentation, other manifestations include the following:
-
Thrombocytopenia (almost 90%) [2]
-
Only hematologic abnormalities (20%) [2]
-
Only infectious manifestations (5%) [2]
-
Only eczema (0%) [2]
-
Autoimmune phenomena [3]
-
Malignancies [3]
See Clinical Presentation for more detail.
Diagnosis
Examination for Wiskott-Aldrich disease includes evaluation for/of the following:
-
Signs of bleeding, infection, malignancy, and atopy
-
General appearance and vital signs
-
Height and weight growth parameters
-
Head and neck assessment
-
Dermatologic assessment
-
Pulmonary assessment
-
Neurologic assessment
Laboratory Tests
Laboratory studies used in the evaluation of Wiskott-Aldrich syndrome include the following:
-
CBC count: Often supports the diagnosis
-
Quantitative serum immunoglobulin levels
-
Functional testing of the humoral and cellular components of the immune system
-
Delayed-type hypersensitivity skin tests
-
Genetic testing
Other tests that may be appropriate, depending on the clinical situation, include the following:
-
Cultures (eg, blood) and sensitivities
-
Renal function tests
-
Hepatic function tests
-
Major histocompatibility tests of the patient, parents, and siblings to determine feasibility for stem cell transplantation
-
Screening of patient and potential donor for infectious agents (eg, HIV, CMV, hepatitis viruses)
-
IL-18 may represent a potential inflammation biomarker in Wiskott-Aldrich syndrome [4]
Imaging studies
Radiography, particularly of the chest, is part of the assessment for new infections. However, CT and MRI studies are not usually utilized for Wiskott-Aldrich syndrome unless stem cell reconstitution procedures have been performed and posttransplantation complications have developed.
Procedures
Consider obtaining a bone marrow biopsy to assist diagnosis in complex cases or to evaluate for hematologic malignancy. However, patients generally do not require bone marrow biopsy.
See Workup for more detail.
Management
Wiskott-Aldrich syndrome has a variable disease severity, depending on the genotype. [5] Accordingly, treatment strategies range from conservative to early definitive intervention, including antibiotics, antivirals, antifungals, chemotherapeutic agents, immunoglobulins, and corticosteroids. Agents are selected based on the patient's clinical presentation and response.
Pharmacotherapy
Medications used in the treatment of Wiskott-Aldrich disease include the following:
-
Antibiotics (eg, amoxicillin, amoxicillin/clavulanate, cefuroxime, ceftriaxone, vancomycin, nafcillin)
-
Inhaled bronchodilators (eg, albuterol, salmeterol, beclomethasone, fluticasone)
-
Hyperimmune globulins (eg, varicella-zoster immune globulin)
-
Immunizations (eg, vaccines, including diphtheria and tetanus toxoids [DT or Td], acellular pertussis, conjugated HIB, conjugated pneumococcal vaccine, unconjugated meningococcal A and C, hepatitis B [HBV], influenza)
-
Corticosteroids (eg, prednisone, methylprednisolone, fluocinolone)
-
Immunoglobulins (eg, immune globulin)
Surgery
Surgical intervention may be necessary for complications of bleeding, such as the following:
-
Neurosurgery if subdural hematoma forms
-
Surgical evacuation of hematomas
-
Surgical intervention to halt blood loss after any minor trauma
-
Splenectomy as an option in cases of coexisting severe thrombocytopenia and frequent bleeding when stem cell reconstitution is not considered
Additional treatments
Supportive care in patients with Wiskott-Aldrich syndrome includes the following:
-
Transfusions of platelets and/or red blood cells
-
Bone marrow transplantation
-
Infusions of intravenous immunoglobulin G
See Treatment and Medication for more detail.
Background
Wiskott-Aldrich syndrome (WAS) was first described by Wiskott in 1937 and was further characterized by Aldrich in 1954. It is a rare X-linked recessive immunodeficiency disorder characterized by the triad of recurrent bacterial sinopulmonary infections, eczema (atopiclike dermatitis), and a bleeding diathesis caused by thrombocytopenia and platelet dysfunction. [6] However, only a third of patients with the syndrome have the classic triad. [7] Almost 90% of patients have manifestations of thrombocytopenia at presentation, 20% have only hematologic abnormalities, 5% have only infectious manifestations, and none have only eczema. [2] WAS platelets are usually smaller than those of idiopathic thrombocytopenia, but a macrothrombocytopenia has been described in WAS. [8] Other symptoms may include autoimmune phenomena and malignancies. [3]
An infant with WAS is seen in the image below.
This 10-month-old infant presented with bloody diarrhea at age 4 months followed by recurrent otitis media infections. A maternal uncle had Wiskott-Aldrich Syndrome (WAS). Note the mild malar eczema and pretibial ecchymoses in this nonambulatory child. His diagnosis was confirmed by immunologic parameters, thrombocytopenia, and low platelet volume.
Wiskott-Aldrich syndrome occurs in males but can occur in females when the X chromosome that contains the functional allele is inactivated, although this is rare. There may be multiple revertant genotypes in patients with Wiskott-Aldrich syndrome. [9]
The gene for the Wiskott-Aldrich syndrome protein (WASp) is localized to Xp11.22-23 and consists of 12 exons that encode a 502 amino acid (53 kD) protein. WASp is a cytosolic protein expressed on all hematopoietic cell lineages and is essential for normal antibody function, T-cell responses, and platelet production. [10] It also regulates actin polymerization, transcription, and a selective, post-transcriptional role in Th2 effector function. [11] About 300 mutations have been found throughout the gene and can include base pair substitutions, insertions, and deletions. These mutations can result in different clinical phenotypes, including classic Wiskott-Aldrich syndrome, X-linked thrombocytopenia, intermittent thrombocytopenia, and neutropenia. [12, 13]
The type of specific mutation, its location within the gene, and its effect on protein expression appear to determine an individual patient's clinical phenotype. [14]
For more information, see Dermatologic Manifestations of Wiskott-Aldrich Syndrome and Wiskott-Aldrich Syndrome.
Pathophysiology
WASP is a key regulator of actin polymerization in hematopoietic cells. As a cytoskeletal regulator, it is necessary for induction of normal immunity. WASp functions as a bridge between signaling and movement of the actin filaments in the cytoskeleton. WASp has several well-defined domains (pleckstrin, cofilin, verprolin, SH3) that are involved in signaling, cell locomotion, and immune synapse formation.
In vitro studies with T cells, platelets, phagocytes, and dendritic cells of patients with Wiskott-Aldrich syndrome reveal defects in the formation of microvilli, filopodia, phagocytic vacuoles, and podosomes, respectively; these structures depend on cytoskeletal reorganization of actin filaments. Researchers also identified many different mutations that interfere with the protein binding to Cdc42 and Rac GTPases, among other binding partners, most of which are involved in regulation of the actin cytoskeleton of lymphocytes. [15, 16] The actin cytoskeleton is responsible for cellular functions, such as growth, endocytosis, exocytosis, and cytokinesis.
Mutations of WASP are located throughout the gene and either inhibit or dysregulate normal WASp function. WASp facilitates the nuclear translocation of nuclear factor kappa-B (NF-kB) and was shown to play an important role in lymphoid development and in the maturation and function of myeloid monocytic cells. In mice, WASp was found to be essential for NF-ATp activation, and for nuclear translocation of p-Erk, Elk1 phosphorylation, and c-fos gene expression in T cells. These defects in mutated forms of WASP are the likely etiology of defective IL-2 expression and T-cell proliferation in Wiskott-Aldrich syndrome. Low T cell numbers resembling T-B+ SCID has been described in WAS. [17]
Clot formation is interrupted by impaired formation of fibrin strands. WASp binds to calcium and integrin binding protein (CIB) on platelets. The complex of CIB and mutated WASp reduces alpha2-beta3 integrin mediated cell adhesion and causes defective platelet aggregation, resulting in bleeding.
Research has shown phenotype-genotype correlation. Classic Wiskott-Aldrich syndrome, X-linked thrombocytopenia, and X-linked neutropenia occurs when WASp is absent, when mutated WASp is expressed, and when missense mutations occur in the Cdc42-binding site, respectively. Although exceptions are noted and although predicting long-term prognosis based on these findings is difficult, this research may lead the way to curative hematopoietic stem cell transplantation and gene therapy. [12] Further research is underway to identify WASp-associated proteins, such as WASp-interacting protein (WIP) and several Wiskott-Aldrich syndrome proteins verprolin homologous (WAVE). [18, 19, 20, 21]
Epidemiology
Frequency
The estimated incidence of Wiskott-Aldrich syndrome in the United States is 1 in 250,000 live male births. [22]
The frequency in the European population has been reported to be similar to that of the United States (1 in 250,000 live male births). A study from Switzerland reported the incidence of Wiskott-Aldrich syndrome is 4.1 cases per 1 million live births. The same study also examined the prevalence of Wiskott-Aldrich syndrome in several national registries (ie, Italy, Japan, Switzerland, Sweden) and found that this condition occurred in 2-8.8% of patients with primary immunodeficiencies. [23] A similar range has been documented in a national registry in Ireland, as well. [24]
Demographics
Wiskott-Aldrich syndrome has been reported in individuals of European, African, and Asian ancestry; however, Blacks and Asians are less likely to be affected. One large series of 301 cases of Wiskott-Aldrich syndrome from 149 families reported that 8 families were black and 4 families were Chicano. [22] Of the 40 families whose ancestry was traced outside North America, 38 emigrated from Europe.
More than 90% of affected patients are male, but females have been reported in the literature. Females typically have no family history. In some cases, females have been shown to have nonrandom inactivation of the X chromosome bearing the functional Wiskott-Aldrich syndrome allele. [25]
Age at presentation ranges from birth to 25 years. In one review, the average age of presentation was 21 months. [2, 22] Male infants present at birth with petechiae and ecchymoses. Infections usually begin in early infancy after maternal immunoglobulin G (IgG) is lost during the first 3 months of life. The frequency of infections usually increase with age. Patients are especially susceptible to encapsulated organisms. Eczema develops during the first year of life and resembles classic atopic dermatitis. Malignancies may occur in children but are more frequent in affected adults. Monoclonal gammopathy is rare and may be of uncertain significance in these children. [26] Lymphomas occur in 26% of patients aged 20 years and older.
Mortality/Morbidity
Morbidity and mortality have gradually improved with better antibiotics, advances in blood banking, better supportive care, and the ability to successfully provide immune reconstitution by stem cell transplantation. Median survival has increased from 8 months in patients born before 1935 to longer than 6 years in patients born after 1964. [22] In one case series, 94 surviving patients ranged in age from 1-35 years, with a median of 11 years; the average age of patients who died was 8 years. [2]
In one study the reported cause of death among patients who did not receive bone marrow transplants were infection (44%), bleeding (23%), or malignancy (26%). [2] Younger patients are more likely to die from bleeding, children are more likely to die from infection, and children and young adults die most often from malignancies. Malignancies may occur in children but are more frequent in affected adults. Lymphomas occur in 26% of patients aged 20 years and older. In one series, 12% of patients developed malignancies, primarily lymphoreticular tumors, and leukemia. In that series, the relative risk of malignancy was more than 100-fold that of normal and the risk increased with age. [22]
The average lifespan for patients who do not receive immune reconstitution is the second to third decade of life, although patients have survived into the fifth decade of life. Following major histocompatibility complex (MHC)–matched stem cell transplantation, the patient who escapes graft versus host disease (GVHD) usually has completely normal immune function and, therefore, has an excellent prognosis for normal survival. [27] Survival rates after stem cell transplant have continued to improve, particularly after more recent emphasis on performing these procedures as soon as possible after diagnosis. [28]
Prognosis
About one fourth of patients who do not receive stem cell reconstitution die from bleeding, another fourth from malignancies, and the remaining 50% from infections. Average age of surviving patients with Wiskott-Aldrich syndrome in 1994 was 11 years, whereas death during the 1960s occurred within 4 years. More recent studies show average age of survival to be around 15 years. Autoimmune disease is a poor prognosis factor in these patients and should be treated promptly. [29]
Hematopoietic stem cell transplantation is the most reliable curative approach for patients with HLA-matched family or unrelated donors. [30, 31] The outlook for successfully transplanted patients is much more optimistic; the first patient to receive complete immunologic reconstitution after a 1968 bone marrow transplantation still survives without immunologic or clinical abnormalities.
Patient Education
As with any patient who has an immune deficiency, the patient and family must seek immediate medical care at the slightest indication of an infection. This issue is critical for the splenectomized patient with Wiskott-Aldrich syndrome who has a high risk of dying from overwhelming postsplenectomy sepsis, usually caused by S pneumoniae infection. Bleeding (eg, epistaxis, into joints, progressive hematomas) must be recognized and treated. Patient and family must be made aware of the risk for complications, including specific autoimmune disorders and malignancies.
An important resource for education and support for patients and families with any primary immunodeficiency disease is the Immune Deficiency Foundation (some states have local chapters).
Immune Deficiency Foundation
25 W Chesapeake Ave, Suite 206
Towson, MD 21204
Consultation calls: 1-877-666-0866
The Jeffrey Modell Foundation also provides educational support and raises funds for research.
The Jeffrey Modell Foundation
747 3rd Avenue
New York, NY 10017
Phone: 1-800-JEFF-844
For patient education resources, see the Skin, Hair, and Nails Center, as well as Eczema.
-
This 10-month-old infant presented with bloody diarrhea at age 4 months followed by recurrent otitis media infections. A maternal uncle had Wiskott-Aldrich Syndrome (WAS). Note the mild malar eczema and pretibial ecchymoses in this nonambulatory child. His diagnosis was confirmed by immunologic parameters, thrombocytopenia, and low platelet volume.
-
This 1-year-old boy was hospitalized because of respiratory syncytial virus bronchiolitis but was noted to have eczema and petechiae (note arrow). His history was significant for a subdural hematoma for which trauma was denied; at that time the platelet count was 212,000. His diagnosis of Wiskott-Aldrich Syndrome (WAS) was confirmed by the detection of a missense mutation (Phe 128 Ser).
Tables
Brand(Manufacturer) |
Manufacturing Process |
pH |
Additives (IVIG products containing sucrose are more often associated with renal dysfunction, acute renal failure, and osmotic nephrosis, particularly with preexisting risk factors [eg, history of renal insufficiency, diabetes mellitus, age >65 y, dehydration, sepsis, paraproteinemia, nephrotoxic drugs].) |
Parenteral Form and Final Concentrations |
IgA Content mcg/mL |
Carimune NF (CSL Behring) |
Kistler-Nitschmann fractionation; pH 4 incubation, nanofiltration |
6.4-6.8 |
6% solution: 10% sucrose, < 20 mg NaCl/g protein |
Lyophilized powder 3%, 6%, 9%, 12% |
Trace |
Flebogamma (Grifols USA) |
Cohn-Oncley fractionation, PEG precipitation, ion-exchange chromatography, pasteurization |
5.1-6 |
Sucrose free, contains 5% D-sorbitol |
Liquid 5% |
< 50 |
Gammagard Liquid 10% (Baxter Bioscience) |
Cohn-Oncley cold ethanol fractionation, cation and anion exchange chromatography, solvent detergent treated, nanofiltration, low pH incubation |
4.6-5.1 |
0.25M glycine |
Ready-for-use Liquid 10% |
37 |
Gamunex (Talecris Biotherapeutics) |
Cohn-Oncley fractionation, caprylate-chromatography purification, cloth and depth filtration, low pH incubation |
4-4.5 |
Contains no sugar, contains glycine |
Liquid 10% |
46 |
Gammaplex (Bio Products) |
Solvent/detergent treatment targeted to enveloped viruses; virus filtration using Pall Ultipor to remove small viruses including nonenveloped viruses; low pH incubation |
4.8-5.1 |
Contains sorbitol (40 mg/mL); do not administer if fructose intolerant |
Ready-for-use solution 5% |
< 10 |
Iveegam EN (Baxter Bioscience) |
Cohn-Oncley fraction II/III; ultrafiltration; pasteurization |
6.4-7.2 |
5% solution: 5% glucose, 0.3% NaCl |
Lyophilized powder 5% |
< 10 |
Polygam S/D Gammagard S/D (Baxter Bioscience for the American Red Cross) |
Cohn-Oncley cold ethanol fractionation, followed by ultracentrafiltration and ion exchange chromatography; solvent detergent treated |
6.4-7.2 |
5% solution: 0.3% albumin, 2.25% glycine, 2% glucose |
Lyophilized powder 5%, 10% |
< 1.6 (5% solution) |
Octagam (Octapharma USA) 9/24/10: Withdrawn from market because of unexplained reports of thromboembolic events |
Cohn-Oncley fraction II/III; ultrafiltration; low pH incubation; S/D treatment pasteurization |
5.1-6 |
10% maltose |
Liquid 5% |
200 |
Panglobulin (Swiss Red Cross for the American Red Cross) |
Kistler-Nitschmann fractionation; pH 4, trace pepsin, nanofiltration |
6.6 |
Per gram of IgG: 1.67 g sucrose, < 20 mg NaCl |
Lyophilized powder 3%, 6%, 9%, 12% |
720 |
Privigen Liquid 10% (CSL Behring) |
Cold ethanol fractionation, octanoic acid fractionation, and anion exchange chromatography; pH 4 incubation and depth filtration |
4.6-5 |
L-proline (~250 mmol/L) as stabilizer; trace sodium; does not contain carbohydrate stabilizers (eg, sucrose, maltose) |
Ready-for-use liquid 10% |
≤ 25 |




