Practice Essentials
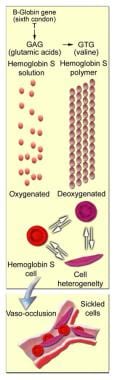
Sickle cell disease (SCD) and its variants are genetic disorders resulting from the presence of a mutated form of hemoglobin, hemoglobin S (HbS) [1, 2] (see the image below). The most common form of SCD found in North America is homozygous HbS disease (HbSS), an autosomal recessive disorder first described by Herrick in 1910. SCD causes significant morbidity and mortality, particularly in people of African and Mediterranean ancestry (see Pathophysiology). Morbidity, frequency of crisis, degree of anemia, and the organ systems involved vary considerably from individual to individual.
Signs and symptoms
Screening for HbS at birth is currently mandatory in the United States. For the first 6 months of life, infants are protected largely by elevated levels of fetal hemoglobin (Hb F). SCD usually manifests early in childhood, in the following ways:
-
Acute and chronic pain: The most common clinical manifestation of SCD is vaso-occlusive crisis; pain crises are the most distinguishing clinical feature of SCD
-
Bone pain: Often seen in long bones of extremities, primarily due to bone marrow infarction
-
Anemia: Universally present, chronic, and hemolytic in nature
-
Aplastic crisis: Serious complication due to infection with parvovirus B19 (B19V)
-
Splenic sequestration: Characterized by the onset of life-threatening anemia with rapid enlargement of the spleen and high reticulocyte count
-
Infection: Organisms that pose the greatest danger include encapsulated respiratory bacteria, particularly Streptococcus pneumoniae; adult infections are predominantly with gram-negative organisms, especially Salmonella
-
Growth retardation, delayed sexual maturation, being underweight
-
Hand-foot syndrome: This is a dactylitis presenting as bilateral painful and swollen hands and/or feet in children
-
Acute chest syndrome: Young children present with chest pain, fever, cough, tachypnea, leukocytosis, and pulmonary infiltrates in the upper lobes; adults are usually afebrile, dyspneic with severe chest pain, with multilobar/lower lobe disease
-
Pulmonary hypertension: Increasingly recognized as a serious complication of SCD
-
Avascular necrosis of the femoral or humeral head: Due to vascular occlusion
-
Central nervous system (CNS) involvement: Most severe manifestation is stroke
-
Ophthalmologic involvement: Ptosis, retinal vascular changes, proliferative retinitis
-
Cardiac involvement: Dilation of both ventricles and the left atrium
-
Gastrointestinal involvement: Cholelithiasis is common in children; liver may become involved
-
Genitourinary involvement: Kidneys lose concentrating capacity; priapism is a well-recognized complication of SCD
-
Dermatologic involvement: Leg ulcers are a chronic painful problem
Approximately half the individuals with homozygous HbS disease experience vaso-occlusive crises. The frequency of crises is extremely variable. Some individuals have as many as 6 or more episodes annually, whereas others may have episodes only at great intervals or none at all. Each individual typically has a consistent pattern for crisis frequency. Triggers of vaso-occlusive crisis include the following:
-
Hypoxemia: May be due to acute chest syndrome or respiratory complications
-
Dehydration: Acidosis results in a shift of the oxygen dissociation curve
-
Changes in body temperature (eg, an increase due to fever or a decrease due to environmental temperature change)
Many individuals with HbSS experience chronic low-level pain, mainly in bones and joints. Intermittent vaso-occlusive crises may be superimposed, or chronic low-level pain may be the only expression of the disease.
See Presentation for more detail.
Diagnosis
SCD is suggested by the typical clinical picture of chronic hemolytic anemia and vaso-occlusive crisis. Electrophoresis confirms the diagnosis with the presence of homozygous HbS and can also document other hemoglobinopathies (eg, HbSC, HbS-beta+ thalassemia).
Laboratory tests used in patients with SCD include the following:
-
Mandatory screening for HbS at birth in the United States; prenatal testing can be obtained via chorionic villus sampling
-
Hemoglobin electrophoresis
-
CBC count with differential and reticulocyte count
-
Serum electrolytes
-
Hemoglobin solubility testing
-
Peripheral blood smear
-
Pulmonary function tests (transcutaneous O 2 saturation)
-
Kidney function (creatinine, BUN, urinalysis)
-
Hepatobiliary function tests, (ALT, fractionated bilirubin)
-
CSF examination: Consider LP in febrile children who appear toxic and in those with neurologic findings (eg, neck stiffness, positive Brudzinski/Kernig signs, focal deficits); consider CT scanning before performing lumbar puncture
-
Blood cultures
-
ABGs
-
Secretory phospholipase A2 (sPLA2)
In one study of 38 asymptomatic children with SCD, investigators found that hypertension and abnormal blood pressure patterns were prevalent in children with SCD. [3] They suggested using 24-hour ambulatory blood pressure monitoring (ABPM) to identify these conditions in young patients. [3]
In the study, 17 patients (43.6%) had ambulatory hypertension, whereas 4 (10.3%) had hypertension on the basis of their clinic blood pressure. Twenty-three patients (59%) had impaired systolic blood pressure dipping, 7 (18%) had impaired diastolic blood pressure dipping, and 5 (13%) had reversed dipping. [3]
Imaging studies
Imaging studies that aid in the diagnosis of sickle cell anemia in patients in whom the disease is suggested clinically include the following:
-
Radiography: Chest x-rays should be performed in patients with respiratory symptoms
-
MRI: Useful for early detection of bone marrow changes due to acute and chronic bone marrow infarction, marrow hyperplasia, osteomyelitis, and osteonecrosis
-
CT scanning: May demonstrate subtle regions of osteonecrosis not apparent on plain radiographs in patients who are unable to have an MRI [4] and to exclude renal medullary carcinoma in patients presenting with hematuria
-
Nuclear medicine scanning: 99mTc bone scanning detects early stages of osteonecrosis; 111In WBC scanning is used for diagnosing osteomyelitis
-
Transcranial Doppler ultrasonography: Can identify children with SCD at high risk for stroke
-
Abdominal ultrasonography: May be used to rule out cholecystitis, cholelithiasis, or an ectopic pregnancy and to measure spleen and liver size
-
Echocardiography: Identifies patients with pulmonary hypertension
-
Transcranial near-infrared spectroscopy or cerebral oximetry: Can be used as a screening tool for low cerebral venous oxygen saturation in children with SCD
See Workup for more detail.
Management
The goals of treatment in SCD are symptom control and management of disease complications. Treatment strategies include the following 7 goals:
-
Management of vaso-occlusive crisis
-
Management of chronic pain syndromes
-
Management of chronic hemolytic anemia
-
Prevention and treatment of infections
-
Management of the complications and the various organ damage syndromes associated with the disease
-
Prevention of stroke
-
Detection and treatment of pulmonary hypertension
Pharmacotherapy
SCD may be treated with the following medications:
-
Antimetabolites: Hydroxyurea
-
Hemoglobin oxygen-affinity modulators (eg, voxelotor)
-
P-selectin inhibitors (eg, crizanlizumab)
-
Gene-editing biologics (ie, exagamglogene autotemcel, lovotibeglogene autotemcel)
-
Opioid analgesics (eg, oxycodone/aspirin, methadone, morphine sulfate, oxycodone/acetaminophen, fentanyl, nalbuphine, codeine, acetaminophen/codeine)
-
Nonsteroidal analgesics (eg, ketorolac, aspirin, acetaminophen, ibuprofen)
-
Tricyclic antidepressants (eg, amitriptyline)
-
Antibiotics (eg, cefuroxime, amoxicillin/clavulanate, penicillin VK, ceftriaxone, azithromycin, cefaclor)
-
Vaccines (eg, pneumococcal, meningococcal, influenza, and recommended scheduled childhood/adult vaccinations)
-
Endothelin-1 receptor antagonists (eg, bosentan)
-
Phosphodiesterase inhibitors (eg, sildenafil, tadalafil)
-
Vitamins (eg, folic acid)
-
L-glutamine
-
Antiemetics (eg, promethazine)
Non-pharmacologic therapy
Other approaches to managing SCD include the following:
-
Stem cell transplantation: Can be curative
-
Transfusions: For sudden, severe anemia due to acute splenic sequestration, parvovirus B19 infection, or hyperhemolytic crises
-
Wound debridement
-
Physical therapy
-
Heat and cold application
-
Acupuncture and acupressure
-
Transcutaneous electric nerve stimulation (TENS)
Combination pharmacotherapy and non-pharmacotherapy
-
Vigorous hydration (plus analgesics): For vaso-occlusive crisis
-
Oxygen, antibiotics, analgesics, incentive spirometry, simple transfusion, and bronchodilators: For treatment of acute chest syndrome
See Treatment and Medication for more detail.
Background
Carriers of the sickle cell trait (ie, heterozygotes who carry one HbS allele and one normal adult hemoglobin [HbA] allele) have some resistance to the often-fatal malaria caused by Plasmodium falciparum. This property explains the distribution and persistence of this gene in the population in malaria-endemic areas. [5, 6, 7]
However, in areas such as the United States, where malaria is not a problem, the trait no longer provides a survival advantage. Instead, it poses the threat of SCD, which occurs in children of carriers who inherit the sickle cell gene from both parents (ie, HbSS).
Although carriers of sickle cell trait do not suffer from SCD, individuals with one copy of HbS and one copy of a gene that codes for another abnormal variant of hemoglobin, such as HbC or Hb beta-thalassemia, have a less severe form of the disease.
Genetics
SCD denotes all genotypes containing at least one sickle gene, in which HbS makes up at least half the hemoglobin present. Major sickle genotypes described so far include the following:
-
HbSS disease or sickle cell anemia (the most common form) - Homozygote for the S globin with usually a severe or moderately severe phenotype and with the shortest survival
-
HbS/b-0 thalassemia - Double heterozygote for HbS and b-0 thalassemia; clinically indistinguishable from sickle cell anemia (SCA)
-
HbS/b+ thalassemia - Mild-to-moderate severity with variability in different ethnicities
-
HbSC disease - Double heterozygote for HbS and HbC characterized by moderate clinical severity
-
HbS/hereditary persistence of fetal Hb (S/HPHP) - Very mild or asymptomatic phenotype
-
HbS/HbE syndrome - Very rare with a phenotype usually similar to HbS/b+ thalassemia
-
Rare combinations of HbS with other abnormal hemoglobins such as HbD Los Angeles, G-Philadelphia, HbO Arab, and others
Sickle cell trait or the carrier state is the heterozygous form characterized by the presence of around 40% HbS, absence of anemia, inability to concentrate urine (isosthenuria), and hematuria. Under conditions leading to hypoxia, it may become a pathologic risk factor.
SCD is the most severe and most common form. Affected individuals present with a wide range of clinical problems that result from vascular obstruction and ischemia. Although the disease can be diagnosed at birth, clinical abnormalities usually do not occur before age 6 months, when functional asplenia develops. Functional asplenia results in susceptibility to overwhelming infection with encapsulated bacteria. Subsequently, other organs are damaged. Typical manifestations include recurrent pain and progressive incremental infarction.
Newborn screening for sickle hemoglobinopathies is mandated in 50 states. Therefore, most patients presenting to the ED have a known diagnosis.
Pathophysiology
HbS arises from a mutation substituting thymine for adenine in the sixth codon of the beta-chain gene, GAG to GTG. This causes coding of valine instead of glutamate in position 6 of the Hb beta chain. The resulting Hb has the physical properties of forming polymers under deoxy conditions. It also exhibits changes in solubility and molecular stability. These properties are responsible for the profound clinical expressions of the sickling syndromes.
Under deoxy conditions, HbS undergoes marked decrease in solubility, increased viscosity, and polymer formation at concentrations exceeding 30 g/dL. It forms a gel-like substance containing Hb crystals called tactoids. The gel-like form of Hb is in equilibrium with its liquid-soluble form. A number of factors influence this equilibrium, including oxygen tension, concentration of Hb S, and the presence of other hemoglobins.
Oxygen tension is a factor in that polymer formation occurs only in the deoxy state. If oxygen is present, the liquid state prevails. Concentration of Hb S is a factor in that gelation of HbS occurs at concentrations greater than 20.8 g/dL (the normal cellular Hb concentration is 30 g/dL). The presence of other hemoglobins is a factor in that normal adult hemoglobin (HbA) and fetal hemoglobin (HbF) have an inhibitory effect on gelation.
These and other Hb interactions affect the severity of clinical syndromes. HbSS produces a more severe disease than sickle cell HbC (HbSC), HbSD, HbSO Arab, and Hb with one normal and one sickle allele (HbSA).
When red blood cells (RBCs) containing homozygous HbS are exposed to deoxy conditions, the sickling process begins. A slow and gradual polymer formation ensues. Electron microscopy reveals a parallel array of filaments. Repeated and prolonged sickling involves the membrane; the RBC assumes the characteristic sickled shape. (See image below.)
After recurrent episodes of sickling, membrane damage occurs and the cells are no longer capable of resuming the biconcave shape upon reoxygenation. Thus, they become irreversibly sickled cells (ISCs). From 5-50% of RBCs permanently remain in the sickled shape.
When RBCs sickle, they gain Na+ and lose K+. Membrane permeability to Ca++ increases, possibly due, in part, to impairment in the Ca++ pump that depends on adenosine triphosphatase (ATPase). The intracellular Ca++ concentration rises to 4 times the reference level. The membrane becomes more rigid, possibly due to changes in cytoskeletal protein interactions; however, these changes are not found consistently. In addition, whether calcium is responsible for membrane rigidity is not clear.
Membrane vesicle formation occurs, and the lipid bilayer is perturbed. The outer leaflet has increased amounts of phosphatidyl ethanolamine and contains phosphatidylserine. The latter may play a role as a contributor to thrombosis, acting as a catalyst for plasma clotting factors. Membrane rigidity can be reversed in vitro by replacing HbS with HbA, suggesting that HbS interacts with the cell membrane.
Interactions with vascular endothelium
Complex multifactorial mechanisms involving endothelial dysfunction underlie the acute and chronic manifestations of SCD. [8] A current model proposes that vaso-occlusive crises in SCD result from adhesive interactions of sickle cell RBCs and leukocytes with the endothelium. [9]
In this model, the endothelium becomes activated by sickle cell RBCs, either directly, through adhesion molecules on the RBC surface, or indirectly through plasma proteins (eg, thrombospondin, von Willebrand factor) that act as a soluble bridge molecule. This leads, sequentiallly, to recruitment of adherent leukocytes, activation of recruited neutrophils and of other leukocytes (eg, monocytes or natural killer T cells), interactions of RBCs with adherent neutrophils, and clogging of the vessel by cell aggregates composed of RBCs, adherent leukocytes, and possibly platelets. [9]
Sickle cells express very late antigen–4 (VLA-4) on the surface. VLA-4 interacts with the endothelial cell adhesive molecule, vascular cell adhesive molecule–1 (VCAM-1). VCAM-1 is upregulated by hypoxia and inhibited by nitric oxide.
Hypoxia also decreases nitric oxide production, thereby adding to the adhesion of sickle cells to the vascular endothelium. Nitric oxide is a vasodilator. Free Hb is an avid scavenger of nitric oxide. Because of the continuing active hemolysis, there is free Hb in the plasma, and it scavenges nitric oxide, thus contributing to vasoconstriction.
In addition to leukocyte recruitment, inflammatory activation of endothelium may have an indispensable role in enhanced sickle RBC–endothelium interactions. Sickle RBC adhesion in postcapillary venules can cause increased microvascular transit times and initiate vaso-occlusion.
Several studies have shown involvement of an array of adhesion molecules expressed on sickle RBCs, including CD36, a-4-ß-1 integrin, intercellular cell adhesion molecule–4 (ICAM-4), and basal cell adhesion molecule (B-CAM). [10] Adhesion molecules (ie, P-selectin, VCAM-1, a-V-ß-3 integrin) are also expressed on activated endothelium. Finally, plasma factors and adhesive proteins (ie, thrombospondin [TSP], von Willebrand factor [vWf], laminin) play an important role in this interaction.
For example, the induction of VCAM-1 and P-selectin on activated endothelium is known to enhance sickle RBC interactions. In addition, a-V-ß-3 integrin is upregulated in activated endothelium in patients with sickle cell disease. a-V-ß-3 integrin binds to several adhesive proteins (TSP, vWf, red-cell ICAM-4, and, possibly, soluble laminin) involved in sickle RBC adhesion, and antibodies to this integrin dramatically inhibit sickle RBC adhesion.
In addition, under inflammatory conditions, increased leukocyte recruitment in combination with adhesion of sickle RBCs may further contribute to stasis.
Sickle RBCs adhere to endothelium because of increased stickiness. The endothelium participates in this process, as do neutrophils, which also express increased levels of adhesive molecules.
Deformable sickle cells express CD18 and adhere abnormally to endothelium up to 10 times more than normal cells, while ISCs do not. As paradoxical as it might seem, individuals who produce large numbers of ISCs have fewer vaso-occlusive crises than those with more deformable RBCs.
Other properties of sickle cells
Sickle RBCs also adhere to macrophages. This property may contribute to erythrophagocytosis and the hemolytic process.
The microvascular perfusion at the level of the pre-arterioles is influenced by RBCs containing Hb S polymers. This occurs at arterial oxygen saturation, before any morphologic change is apparent.
Hemolysis is a constant finding in sickle cell syndromes. Approximately one third of RBCs undergo intravascular hemolysis, possibly due to loss of membrane filaments during oxygenation and deoxygenation. The remainder hemolyze by erythrophagocytosis by macrophages. This process can be partially modified by Fc (crystallizable fragment) blockade, suggesting that the process can be mediated by immune mechanisms.
Sickle RBCs have increased immunoglobulin G (IgG) on the cell surface. Vaso-occlusive crisis is often triggered by infection. levels of fibrinogen, fibronectin, and D-dimer are elevated in these patients. Plasma clotting factors likely participate in the microthrombi in the pre-arterioles.
Development of clinical disease
Although hematologic changes indicative of SCD are evident as early as the age of 10 weeks, symptoms usually do not develop until the age of 6-12 months because of high levels of circulating fetal hemoglobin. After infancy, erythrocytes of patients with sickle cell anemia contain approximately 90% hemoglobin S (HbS), 2-10% hemoglobin F (HbF), and a normal amount of minor fraction of adult hemoglobin (HbA2). Adult hemoglobin (HbA), which usually gains prominence at the age of 3 months, is absent.
The physiological changes in RBCs result in a disease with the following cardinal signs:
- Hemolytic anemia
- Painful vaso-occlusive crisis
- Multiple organ damage from microinfarcts, including heart, skeleton, spleen, and central nervous system
Silent cerebral infarcts are associated with cognitive impairment in SCD. These infarcts tend to be located in the deep white matter where cerebral blood flow is low. [11] However, cognitive impairment, particularly slower processing speed, may occur independent of the presence of infarction and may worsen with age. [12]
Musculoskeletal manifestations
The skeletal manifestations of sickle cell disease result from changes in bone and bone marrow caused by chronic tissue hypoxia, which is exacerbated by episodic occlusion of the microcirculation by the abnormal sickle cells. The main processes that lead to bone and joint destruction in sickle cell disease are as follows:
-
Infarction of bone and bone marrow
-
Compensatory bone marrow hyperplasia
-
Secondary osteomyelitis
-
Secondary growth defects
When the rigid erythrocytes jam in the arterial and venous sinusoids of skeletal tissue, the result is intravascular thrombosis, which leads to infarction of bone and bone marrow. Repeated episodes of these crises eventually lead to irreversible bone infarcts and osteonecrosis, especially in weight-bearing areas. These areas of osteonecrosis (avascular necrosis/aseptic necrosis) become radiographically visible as sclerosis of bone with secondary reparative reaction and eventually result in degenerative bone and joint destruction.
Infarction tends to occur in the diaphyses of small tubular bones in children and in the metaphyses and subchondrium of long bones in adults. Because of the anatomic distribution of the blood vessels supplying the vertebrae, infarction affecting the central part of the vertebrae (fed by a spinal artery branch) results in the characteristic H vertebrae of sickle cell disease. The outer portions of the plates are spared because of the numerous apophyseal arteries.
Osteonecrosis of the epiphysis of the femoral head is often bilateral and eventually progresses to collapse of the femoral heads. This same phenomenon is also seen in the humeral head, distal femur, and tibial condyles.
Infarction of bone and bone marrow in patients with sickle cell disease can lead to the following changes (see images below):
-
Osteolysis (in acute infarction)
-
Osteonecrosis (avascular necrosis/aseptic necrosis)
-
Articular disintegration
-
Myelosclerosis
-
Periosteal reaction (unusual in the adult)
-
H vertebrae (steplike endplate depression; also known as the Reynold sign or codfish vertebrae)
-
Dystrophic medullary calcification
-
Bone-within-bone appearance
-
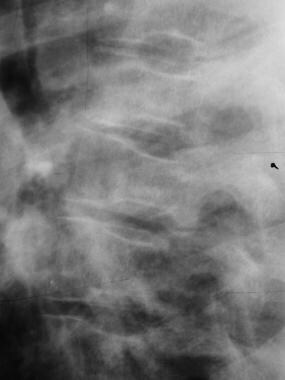 Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
 Skeletal sickle cell anemia. Bone-within-bone appearance. Following multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex.
Skeletal sickle cell anemia. Bone-within-bone appearance. Following multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex.


The shortened survival time of the erythrocytes in sickle cell anemia (10-20 days) leads to a compensatory marrow hyperplasia throughout the skeleton. The bone marrow hyperplasia has the resultant effect of weakening the skeletal tissue by widening the medullary cavities, replacing trabecular bone and thinning cortices.
Deossification due to marrow hyperplasia can bring about the following changes in bone:
-
Decreased density of the skull
-
Decreased thickness of outer tble of skull due to widening of diploe
-
Hair on-end striations of the calvaria
-
Osteoporosis sometimes leading to biconcave vertebrae, coarsening of trabeculae in long and flat bones, and pathologic fractures
Patients with sickle cell disease can have a variety of growth defects due to the abnormal maturation of bone. The following growth defects are often seen in sickle cell disease:
-
Bone shortening (premature epiphyseal fusion)
-
Epiphyseal deformity with cupped metaphysis
-
Peg-in-hole defect of distal femur
-
Decreased height of vertebrae (short stature and kyphoscoliosis)
Go to Skeletal Sickle Cell Anemia for complete information on this topic.
SCD can result in significant skeletal muscle remodeling and reduced muscle functional capacities, which contribute to exercise intolerance and poor quality of life. [13] In addition, changes in muscle and joints can result in altered posture and impaired balance control. [14]
Renal manifestations
Renal manifestations of SCD range from various functional abnormalities to gross anatomic alterations of the kidneys. See Renal Manifestations of Sickle Cell Disease for more information on this topic.
Splenic manifestations
The spleen enlarges in the latter part of the first year of life in children with SCD. Occasionally, the spleen undergoes a sudden very painful enlargement due to pooling of large numbers of sickled cells. This phenomenon is known as splenic sequestration crisis.
The spleen undergoes repeated infarction, aided by low pH and low oxygen tension in the sinusoids and splenic cords. Despite being enlarged, its function is impaired, as evidenced by its failure to take up technetium during nuclear scanning.
Over time, the spleen becomes fibrotic and shrinks. This is, in fact, an autosplenectomy. The nonfunctional spleen is a major contributor to the immune deficiency that exists in these individuals. Failure of opsonization and an inability to deal with infective encapsulated microorganisms, particularly Streptococcus pneumoniae, ensue, leading to an increased risk of sepsis in the future.
Chronic hemolytic anemia
SCD is a form of hemolytic anemia, with red cell survival of around 10-20 days. Approximately one third of the hemolysis occurs intravascularly, releasing free hemoglobin (plasma free hemoglobin [PFH]) and arginase into plasma. PFH has been associated with endothelial injury including scavenging nitric oxide (NO), proinflammatory stress, and coagulopathy, resulting in vasomotor instability and proliferative vasculopathy.
A hallmark of this proliferative vasculopathy is the development of pulmonary hypertension in adulthood. Plasma arginase degrades arginine, the substrate for NO synthesis, thereby limiting the expected compensatory increase in NO production and resulting in generation of oxygen radicals. Plasma arginase is also associated with pulmonary hypertension and risk of early mortality.
Infection
Life-threatening bacterial infections are a major cause of morbidity and mortality in patients with SCD. Recurrent vaso-occlusion induces splenic infarctions and consequent autosplenectomy, predisposing to severe infections with encapsulated organisms (eg, Haemophilus influenzae, Streptococcus pneumoniae).
Lower serum immunoglobulin M (IgM) levels, impaired opsonization, and sluggish alternative complement pathway activation further increase susceptibility to other common infectious agents, including Mycoplasma pneumoniae, Salmonella typhimurium, Staphylococcus aureus, and Escherichia coli. Common infections include pneumonia, bronchitis, cholecystitis, pyelonephritis, cystitis, osteomyelitis, meningitis, and sepsis.
Pneumococcal sepsis continues to be a major cause of death in infants in some countries. Parvovirus B19 infection causes aplastic crises.
Etiology
SCD originated in West Africa, where it has the highest prevalence. It is also present to a lesser extent in India and the Mediterranean region. DNA polymorphism of the beta S gene suggests that it arose from five separate mutations: four in Africa and one in India and the Middle East. The most common of these is an allele found in Benin in West Africa. The other haplotypes are found in Senegal and Bantu, Africa, as well as in India and the Middle East.
The HbS gene, when present in homozygous form, is an undesirable mutation, so a selective advantage in the heterozygous form must account for its high prevalence and persistence. Malaria is possibly the selecting agent because a concordance exists between the prevalence of malaria and Hb S. Sickling might protect a person from malaria by either (1) accelerating sickling so that parasitized cells are removed or (2) making it more difficult for the parasite to metabolize or to enter the sickled cell. While children with sickle cell trait Hb SA seem to have a milder form of falciparum malaria, those with homozygous Hb S have a severe form that is associated with a very high mortality rate.
The sickling process that prompts a crisis may be precipitated by multiple factors. Local tissue hypoxia, dehydration secondary to a viral illness, or nausea and vomiting, all of which lead to hypertonicity of the plasma, may induce sickling. Any event that can lead to acidosis, such as infection or extreme dehydration, can cause sickling. More benign factors and environmental changes, such as fatigue, exposure to cold, and psychosocial stress, can elicit the sickling process. A specific cause is often not identified.
Vaso-occlusive crises are often precipitated by the following:
-
Cold weather (due to vasospasm)
-
Hypoxia (eg, flying in unpressurized aircraft)
-
Infection
-
Dehydration (especially from exertion or during warm weather)
-
Acidosis
-
Alcohol intoxication
-
Emotional stress
-
Pregnancy
Data also suggest a role for exertional stress, particularly when compounded with heat and hypovolemia.
Aplastic crises are often preceded by the following:
-
Infection with parvovirus B19
-
Folic acid deficiency
-
Ingestion of bone marrow toxins (eg, phenylbutazone)
Acute chest syndrome has been linked to the following:
-
Fat embolism
-
Infections
-
Pain episodes
-
Asthma. [15]
Epidemiology
SCD is present mostly in blacks. It also is found, with much less frequency, in eastern Mediterranean and Middle East populations. Individuals of Central African Republic descent are at an increased risk for overt renal failure.
United States statistics
The sickle gene is present in approximately 8% of black Americans. The expected prevalence of sickle cell anemia in the United States is 1 in 625 persons at birth. The actual prevalence is less because of early mortality. More than 2 million people in the United States, nearly all of them of African American ancestry, carry the sickle gene. More than 30,000 patients have homozygous HbS disease.
The following statistics are available from the Centers for Disease Control and Prevention and the National Institutes of Health [16, 17] :
-
Sickle cell anemia is the most common inherited blood disorder in the United States
-
In the United States, approximately 100,000 people have SCD
-
SCD occurs in about 1 out of every 365 Black or African-American births
-
SCD occurs in about 1 of every 16,300 Hispanic-American births
-
Approximately 1 in 13 Blacks or African Americans has sickle cell trait
Risk of chronic kidney disease (CKD) is increased in SCD, with one study showing an almost 30% prevalence of baseline CKD in a cohort with mean age 31.6 years, increasing to 41.8% over 5 years. Among US sickle cell trait carriers of African or Hispanic ancestry, the risk of CKD is about 1.5-2–fold higher than in noncarriers. [18] The prevalence of sickle cell trait is twice as high in African Americans with end-stage renal disease compared with the general African-American population (15% versus 7%, P < 0.001). [19]
Certain gene variants have been linked with increased risk of kidney disease in SCD. In particular, patients who carry APOL1 G1 and G2 risk variants (which confer protection against trypanosomiasis) have a 7-fold higher risk of CKD progression and a 7-30–fold greater risk of kidney failure. [20]
International statistics
In several sections of Africa, the prevalence of sickle cell trait (heterozygosity) is as high as 30%. Although the disease is most frequently found in sub-Saharan Africa, it is also found in some parts of Sicily, Greece, southern Turkey, and India, all of which have areas in which malaria is endemic.
The mutation that results in HbS is believed to have originated in several locations in Africa and India. Its prevalence varies but is high in these countries because of the survival advantage to heterozygotes in regions of endemic malaria. As a result of migration, both forced and voluntary, it is now found worldwide.
Sex distribution
The male-to-female ratio is 1:1. No sex predilection exists, since sickle cell anemia is not an X-linked disease.
Although no particular gender predilection has been shown in most series, analysis of the data from the US Renal Data System demonstrated marked male predominance of sickle cell nephropathy in affected patients. [21]
Clinical characteristics at different ages
Although hematologic changes indicative of the disorder are evident as early as the age of 10 weeks, clinical characteristics of SCD generally do not appear until the second half of the first year of life, when fetal Hb levels decline sufficiently for abnormalities caused by HbS to manifest. SCD then persists for the entire lifespan. After age 10 years, rates of painful crises decrease, but rates of complications increase.
The median age at the time of renal failure in patients with SCD is 23.1 years, the median survival time after the diagnosis of ESRD is about 4 years, and the median age of death is 27 years, despite dialysis treatment. [22]
Prognosis
Because SCD is a lifelong disease, prognosis is guarded. The goal is to achieve a normal life span with minimal morbidity. As therapy improves, the prognosis also improves. Morbidity is highly variable in patients with SCD, partly depending on the level of HbF. Nearly all individuals with the condition are affected to some degree and experience multiple organ system involvement. Patients with Hb SA are heterozygous carriers and essentially are asymptomatic.
Vaso-occlusive crisis and chronic pain are associated with considerable economic loss and disability. Repeated infarction of joints, bones, and growth plates leads to aseptic necrosis, especially in weightbearing areas such as the femur. This complication is associated with chronic pain and disability and may require changes in employment and lifestyle.
Prognostic factors in SCD
The following prognostic factors have been identified as predictors of an adverse outcome [23] :
-
Hand-foot syndrome (dactylitis) in infants younger than 1 year
-
Hb level of less than 7 g/dL
-
Leukocytosis in the absence of infection
Hand-foot syndrome, which affects children younger than 5 years, has proved a strong predictor of overall severity (ie, death, risk of stroke, high pain rate, recurrent acute chest syndrome). Those that have an episode before age 1 year are at high risk of a severe clinical course. The risk is further increased if the child's baseline hemoglobin level is less than 7 g/dL or the baseline WBC count is elevated.
Pregnancy in SCD
Pregnancy represents a special area of concern. The high rate of fetal loss is due to spontaneous abortion. Placenta previa and abruption are common due to hypoxia and placental infarction. At birth, the infant often is premature or has low birth weight.
Mortality in SCD
Mortality is high, especially in the early childhood years. Since the introduction of widespread penicillin prophylaxis and pneumococcal vaccination, a marked reduction has been observed in childhood deaths. The leading cause of death is acute chest syndrome. Children have a higher incidence of acute chest syndrome but a lower mortality rate than adults; the overall death rate from acute chest syndrome is 1.8% and 4 times higher in adults than in children. Causes of death are pulmonary embolism and infection.
In the Dallas newborn cohort, estimated survival at 18 years was 94%. In a recent neonatal United Kingdom cohort followed in a hospital and community-based program including modern therapy with transcranial Doppler ultrasonography (TCD) screening, the estimated survival of HbSS children at 16 years was 99%. Data from the 1995 cooperative study of SCD (CSSCD) suggested that the median survival for individuals with SCD was 48 years for women and 42 years for men. [24] This life expectancy was considerably lower than that for African Americans who do not have SCD.
In Africa, available mortality data are sporadic and incomplete. Many children are not diagnosed, especially in rural areas, and death is often attributed to malaria or other comorbid conditions.
Data from Quinn et al in 2004 suggest that mortality from SCD has improved over the past 30 years. [25] In earlier reports, approximately 50% of patients did not survive beyond age 20 years, and most did not survive to age 50 years.
In one study, the median survival time in patients with SCD after the diagnosis of ESRD was about 4 years, and the median age of death after diagnosis was 27 years, despite dialysis treatment. [22]
The cooperative study of SCD (CSSCD) estimated that the median survival for individuals with SS was 48 years for women and 42 years for men. [24] In the Dallas newborn cohort, estimated survival at 18 years was 94%. In a recent neonatal United Kingdom cohort followed in a hospital and community-based program including modern therapy with TCD screening, the estimated survival of HbSS children at 16 years was 99%.
This significant increase in life expectancy and survival of patients with SCD has been achieved thanks to early detection and introduction of disease-modifying therapies. Neonatal screening, penicillin prophylaxis for children, pneumococcal immunization, red cell transfusion for selected patients and chelation therapy, hydroxyurea therapy, parental and patient education and, above all, treatment in comprehensive centers have all likely contributed to this effect on longevity.
However, as the population of patients with SCD grows older, new chronic complications are appearing. Pulmonary hypertension is emerging as a relatively common complication and is one of the leading causes of morbidity and mortality in adults with SCD. [26]
A study of 398 outpatients with SCD in France found that the prevalence of pulmonary hypertension confirmed by right heart catheterization was 6%; echocardiography alone had a low positive predictive value for pulmonary hypertension. [27]
Patient Education
Patients must be educated about the nature of their disease. They must be able to recognize the earliest signs of a vaso-occlusive crisis and seek help, treat all febrile illness promptly, and identify environmental hazards that may precipitate a crisis. Reinforcement should occur incrementally during the course of ongoing care.
Patients or parents should be instructed on how to palpate the abdomen to detect splenic enlargement, and the importance of observation for pallor, jaundice, and fever. Teach patients to seek medical care in certain situations, including the following:
-
Persistent fever (>38.3°C)
-
Chest pain, shortness of breath, nausea, and vomiting
-
Abdominal pain with nausea and vomiting
-
Persistent headache not experienced previously
Patients should avoid the following:
-
Alcohol
-
Nonprescribed prescription drugs
-
Cigarettes, marijuana, and cocaine
-
Seeking care in multiple institutions
Families should be educated on the importance of hydration, diet, outpatient medications, and immunization protocol. Emphasize the importance of prophylactic penicillin. Patients on hydroxyurea must be educated on the importance of regular follow-up with blood counts.
Patients (including asymptomatic heterozygous carriers) should understand the genetic basis of the disease, be educated about prenatal diagnosis, and know that genetic counseling is available. Genetic testing can identify parents at risk for having a child with sickle cell disease.
If both parents have the sickle cell trait, the chance that a child will have sickle cell disease is 25%. If one parent is carrying the trait and the other actually has disease, the odds increase to 50% that their child will inherit the disease. Screening and genetic counseling theoretically have the potential to drastically reduce the prevalence of SCD. This promise has not been realized. Some authors have recommended emergency department screening or referral for patients unaware of their status as a possible heterozygote. [28]
Families should be encouraged to contact community sickle cell agencies for follow-up information, new drug protocols, and psychosocial support. Families should also follow the advances of gene therapy, bone marrow transplantation, and the usage of cord blood stem cells.
For patient education information, see Sickle Cell Disease. In addition, the Centers for Disease Control and Prevention offers a range of patient-centered materials and multimedia on SCD. [29]
-
Molecular and cellular changes of hemoglobin S.
-
Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
-
Peripheral blood with sickled cells at 400X magnification. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Peripheral blood smear with sickled cells at 1000X magnification. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Peripheral blood smear with Howell-Jolly body, indicating functional asplenism. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Effects of therapy with hydroxyurea.
-
Skeletal sickle cell anemia. Bone-within-bone appearance. Following multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex.
-
A 12-year-old boy with HgbSS disease presents to the pediatric emergency department after his mother tried to wake him for school this morning and noted altered mental status, left-sided gaze paralysis with his head tilted to the left, and flaccid paralysis of the right arm and leg. A CT scan of the brain was obtained immediately.
-
Embolic stroke of the left middle cerebral artery. SCD is the most common cause of stroke in children and one of the most devastating complications of SCD. Clinically overt strokes are typically due to embolism of the intracranial internal carotid artery and proximal middle cerebral artery (MCA), while "silent strokes" more typically occur in the smaller lacunar and penetrating arteries. As RBCs undergo sickling and hemolysis within the cerebral circulation, they adhere to the vascular endothelium and promote a hypercoaguable state and fuel thromboembolism formation. Treatment options include prophylactic therapy with hydroxyurea to promote HgbF concentrations and monitoring via transcranial Doppler to evaluate MCA blood flow velocity. Children found to have high velocities are at increased risk for stroke and commonly receive RBC transfusions to decrease the concentration of HgbS.
-
The spleen enlarges during the first year of life in SCD, as it becomes congested with trapped slow-flowing sickled cells within the splenic sinuses and reticuloendothelial system. The histology image shown demonstrates splenic congestion from sequestered sickled RBCs (arrows). Microvascular occlusions produce chronic tissue hypoxia and microinfarctions. Over time, fibrosis induces autosplenectomy. With functional asplenia, patients are particularly susceptible to infection by the encapsulated organisms Streptococcus pneumoniae and Haemophilus influenzae. Vaccination and prophylactic daily penicillin throughout childhood are mainstays of treatment to prevent sepsis and meningitis
-
Splenic sequestration is an important cause of morbidity that occurs when sudden splenic pooling of blood within the reticuloendothelial system causes an acute drop in circulatory volume and shock-like symptoms (hypotension, tachycardia) with a rigid distended abdomen. It is an acute emergency and can be fatal in 1-2 hours secondary to circulatory hypovolemia. Treatment is with volume resuscitation and blood transfusion. The CT image shown demonstrates splenomegaly with a mass-like process (arrows) from splenic sequestration.
-
Patients with SCD are also at increased risk of developing pulmonary arterial hypertension (PAH). The etiology is most likely multifactorial but likely related to increased cardiac output secondary to underlying chronic anemia. Impedance to the elevated blood flow will cause further dilation and increase in pulmonary pressures. Postsickling changes including interstitial fibrosis secondary to vaso-occlusive crisis of ACS and hypoperfusion with resultant hypoxia of the pulmonary vascular beds are both proposed offenders inciting further dilation and elevation of pulmonary pressures. A pulmonary arteriogram depicting the markedly dilated vascular supply of the lungs seen in PAH is shown.
-
Proliferative sickle cell retinopathy. Sickle cell retinopathy is believed to be vaso-occlusion of peripheral arterioles of the retina leading to retinal hypoxia, ischemia, and infarction. New vessels then form at the junction of the vascular and avascular areas of retina. This neovascularization of retinal tissue and resultant traction of fibrovascularization places patients at risk for vitreous hemorrhage (arrows) and retinal detachment. Another common ocular manifestation is hyphema. Anterior chamber bleeding occurs spontaneously, but sickled erythrocytes obstruct the trabecular meshwork leading to significant elevations of intraocular pressure. Patients are particularly susceptible to glaucomatous optic nerve damage from even mildly elevated intraocular pressures. Pressures greater than 36 mm Hg for 24 hours are an indication for surgical drainage in both SCD and sickle cell trait, regardless of the size of hyphema. Image courtesy of the National Eye Institute, National Institutes of Health (NEI/NIH).
-
A 19-year-old man with known HgbSS disease presents because his girlfriend reports his eyes are yellow. He has no complaints. Physical exam is notable for mild abdominal pain, but is otherwise within normal limits. What imaging test is warranted for this work-up? The image shown is of a male child with similar symptoms. Image courtesy of Wikimedia Commons.
-
Right upper quadrant ultrasoundChronic hemolysis of sickled cells in HgbSS disease and high heme turnover yields hyperbilirubinemia and is associated with increased formation of bile stones. Stone formation occurs as substances in bile reach concentrations that approach the limits of their solubility. As saturation levels are reached, crystals precipitate, become trapped in mucus, and produce sludge (shown). Over time, the crystals aggregate and form stones. Occlusion of the biliary tree by sludge and/or stones produces clinical disease, typically right upper quadrant pain. The scleral icterus seen in the image of the previous slide is most likely secondary to the elevated circulating levels of bilirubin as a result of an acute hemolytic event (such as an acute vaso-occlusive crisis).
-
Renal papillary necrosisThe microvascular beds of the renal parenchyma are susceptible to sickling and vaso-occlusive crisis because of their inherent low-oxygen and high-osmolarity state. Depending on the location of occlusion, symptoms vary from a decreased ability to concentrate urine (yielding nocturia and polyuria), a disruption of exchange mechanisms (yielding hyperkalemia) or hematuria, which further damages renal tubules. In renal papillary necrosis, repeated vascular occlusion infarcts the renal medullary pyramids and papillae. This causes sloughing of papillae, which obstructs the urinary tract. Treatment options include hydration, high-dose antibiotics for resulting pyelonephritis, and possible percutaneous nephrostomy tube or invasive retrieval of sloughed papillae in acute urinary obstruction. The intravenous pyelogram demonstrates the "egg-in-a-cup" appearance of sloughed renal papillae (arrows) secondary to renal papillary necrosis.
-
Skeletal sickle cell anemia. Hand-foot syndrome. Soft tissue swelling with periosteal new-bone formation and a moth-eaten lytic process at the proximal aspect of the fourth phalanx.
-
Skeletal sickle cell anemia. Advanced dactylitis. Lytic processes are present at the first and fifth metacarpals, along with periostitis, which is most prominent in the third metacarpal.
-
Skeletal sickle cell anemia. Expanded medullary cavity. The diploic space is markedly widened due to marrow hyperplasia. Trabeculae are oriented perpendicular to the inner table, giving a hair-on-end appearance.
-
Skeletal sickle cell anemia. Detailed view of the expanded medullary cavity in the same patient as in the previous image.
-
Skeletal sickle cell anemia. Osteonecrosis. Image shows flattening of the femoral heads with a mixture of sclerosis and lucency characteristic of osteonecrosis.
-
Skeletal sickle cell anemia. Osteonecrosis. Detail of the right hip.
-
Skeletal sickle cell anemia. Osteonecrosis. Detail of the left hip.
-
Skeletal sickle cell anemia. Bone infarct. Image shows patchy sclerosis of the humeral head and shaft representing multiple prior bone infarcts.
-
Skeletal sickle cell anemia. Chronic infarcts and secondary osteoarthritis. Image shows advanced changes of irregular sclerosis and lucency on both sides of the knee joint reflecting numerous prior infarcts. The joint surfaces are irregular and the cartilages are narrowed due to secondary osteoarthritis.
-
Skeletal sickle cell anemia. Osteonecrosis. Coronal T1-weighted MRI shows a slightly flattened femoral head with a serpentine margin of low signal intensity around an area of ischemic marrow with signal intensity similar to that of fat.
-
Skeletal sickle cell anemia. Osteonecrosis in the same patient as in the previous image. Coronal T2-weighted MRI shows a serpentine area of low signal intensity and additional focal areas of abnormal low signal intensity in the femoral head; these findings reflect collapse of bone and sclerosis.
-
Skeletal sickle cell anemia. Osteomyelitis. CT scan in a soft tissue window demonstrates a large abscess in the left thigh encircling the femur, with hypoattenuating pus surrounded by a rim of vivid enhancement.
-
Skeletal sickle cell anemia. Osteomyelitis and bone-within-bone. Bone-window CT scan in the same patient as in the previous image shows a bone-within-bone appearance (concentric rings of cortical bone) in the right femur. On the left, a sinus tract (cloaca) traverses the lateral aspect of the femoral cortex, and a small, shardlike sequestrum is present deep to the sinus tract.
-
Skeletal sickle cell anemia. Bone scan of bone infarct shows an area of increased uptake in the distal femoral metaphysis corresponding to the infarct demonstrated on the previous plain radiograph.
Tables
Tests |
Age |
Frequency |
CBC count with WBC differential, reticulocyte count |
3-24 mo >24 mo |
every 3 mo every 6 mo |
Percent Hb F |
6-24 mo >24 mo |
every 6 mo annually |
Renal function (creatinine, BUN, urinalysis) |
≥ 12 mo |
annually |
Hepatobiliary function (ALT, fractionated bilirubin) |
≥ 12 mo |
annually |
Pulmonary function (transcutaneous O2 saturation) |
≥ 12 mo |
every 6 mo |

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Approach Considerations
- Baseline Blood Study Abnormalities
- Suggested Routine Clinical Laboratory Evaluations
- Laboratory Studies in the Ill Child
- Additional Tests
- Radiography
- Magnetic Resonance Imaging
- Nuclear Medicine Scans
- Transcranial Doppler Ultrasonography
- Abdominal Ultrasonography
- Echocardiography
- Show All
- Treatment
- Approach Considerations
- Hydroxyurea Therapy
- Transfusion
- Management of Ophthalmic Manifestations
- Vaso-Occlusive Crisis Management
- Control of Acute Pain
- Treatment of Acute Chest Syndrome
- Control of Chronic Pain
- Management of Chronic Anemia
- Prevention and Treatment of Infections
- Treatment of Gallstones
- Treatment of Priapism
- Treatment of Leg Ulcers
- Stroke Prevention
- Treatment of Pulmonary Hypertension
- Sickle Cell Nephropathy
- Treatment of Other Complications
- Gene Therapy
- Stem Cell Transplantation
- Diet and Activity Restrictions
- Investigational Treatments
- Consultations
- Long-Term Monitoring
- Show All
- Guidelines
- Medication
- Medication Summary
- Antimetabolites
- Hemoglobin Oxygen-Affinity Modulators
- P-Selectin Inhibitor
- Gene Therapies, Hematologics
- Opioid Analgesics
- Nonsteroidal Analgesics
- Tricyclic Antidepressants
- Vitamins
- Nutritionals
- Antibiotics
- Phosphodiesterase Type 5 Inhibitors
- Endothelin Receptor Antagonists
- Iron Chelators
- Antiemetics
- Adrenergic Agonists
- Vaccines
- Show All
- Questions & Answers
- Media Gallery
- Tables
- References






