Background
Respiratory distress syndrome, also known as hyaline membrane disease, occurs almost exclusively in premature infants. The incidence and severity of respiratory distress syndrome are related inversely to the gestational age of the newborn infant. (See Etiology and Epidemiology.)
Enormous strides have been made in understanding the pathophysiology and management of respiratory distress syndrome, leading to improvements in morbidity and mortality in infants with the condition. Advances include the following (see Treatment and Medication):
-
The use of antenatal steroids to enhance pulmonary maturity
-
Appropriate resuscitation facilitated by placental transfusion and immediate use of continuous positive airway pressure (CPAP) for alveolar recruitment
-
Early administration of surfactant
-
The use of gentler modes of ventilation, including early use of "bubble" nasal CPAP to minimize damage to the immature lungs
-
Supportive therapies, such as the diagnosis and management of patent ductus arteriosus (PDA), fluid and electrolyte management, trophic feeding and nutrition, and the use of prophylactic fluconazole
These therapies have also resulted in the survival of extremely premature infants, some of who continue to be ill with complications of prematurity. (See the image below.)
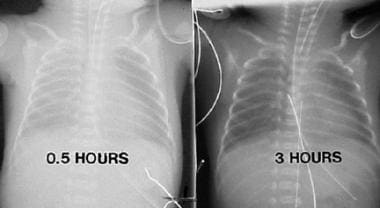 Chest radiographs in a premature infant with respiratory distress syndrome before and after surfactant treatment. Left: Initial radiograph shows poor lung expansion, air bronchogram, and reticular granular appearance. Right: Repeat chest radiograph obtained when the neonate is aged 3 hours and after surfactant therapy demonstrates marked improvement.
Chest radiographs in a premature infant with respiratory distress syndrome before and after surfactant treatment. Left: Initial radiograph shows poor lung expansion, air bronchogram, and reticular granular appearance. Right: Repeat chest radiograph obtained when the neonate is aged 3 hours and after surfactant therapy demonstrates marked improvement.
Complications
Although reduced, the incidence and severity of complications of respiratory distress syndrome can result in clinically significant morbidities. Sequelae of respiratory distress syndrome include the following (see Prognosis, Presentation, and Workup):
-
Septicemia
-
Bronchopulmonary dysplasia (BPD)
-
Patent ductus arteriosus (PDA)
-
Pulmonary hemorrhage
-
Apnea/bradycardia
-
Intraventricular hemorrhage (IVH)
-
Periventricular leukomalacia (PVL) - With associated neurodevelopmental and audiovisual handicaps
Strategic goals include focusing direct attention on anticipating and minimizing these complications and preventing premature delivery whenever possible. (See the diagram below.)
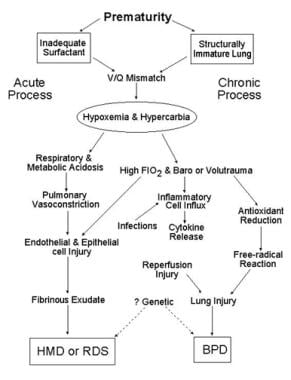 Schematic outlines the pathology of respiratory distress syndrome (RDS). Infants may recover completely or develop chronic lung damage, resulting in bronchopulmonary dysplasia (BPD). FiO2 = fraction of inspired oxygen; HMD = hyaline membrane disease; V/Q = ventilation perfusion.
Schematic outlines the pathology of respiratory distress syndrome (RDS). Infants may recover completely or develop chronic lung damage, resulting in bronchopulmonary dysplasia (BPD). FiO2 = fraction of inspired oxygen; HMD = hyaline membrane disease; V/Q = ventilation perfusion.
Surfactant formation and physiology
Surfactant is a complex lipoprotein (see the image below) composed of six phospholipids and four apoproteins. Surfactant recovered by alveolar wash from most mammals contains 70-80% phospholipids, 8-10% protein, and 10% neutral lipids, primarily cholesterol. Dipalmitoyl phosphatidylcholine (DPPC), or lecithin, is functionally the principle phospholipid. Phosphatidylglycerol makes up 4-15% of the phospholipids; although it is a marker for lung maturity, it is not necessary for normal lung function.
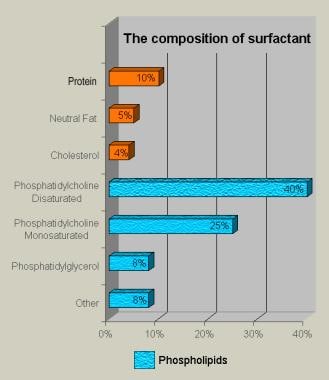 Bar chart demonstrates the composition of lung surfactant. About 1% of the 10% protein component comprises surfactant apoproteins; the remaining proteins are derived from alveolar exudate.
Bar chart demonstrates the composition of lung surfactant. About 1% of the 10% protein component comprises surfactant apoproteins; the remaining proteins are derived from alveolar exudate.
Among the four surfactant apoproteins identified, surfactant protein B (SP-B) and SP-C are two small hydrophobic proteins that make up 2-4% of the surfactant mass and are present in commercially available surfactant preparations. SP-B and SP-C work in concert to facilitate rapid adsorption and spreading of DPPC as a monolayer to lower the surface tension at the alveolar air-fluid interface in vivo during expiration, thus preventing atelectasis.
The SP-B gene is on human chromosome 2, and its primary translation product is 40 kd, which is clipped to become an 8-kd protein in the type II cells before entering lamellar bodies to be cosecreted with phospholipids. The SP-C gene is on chromosome 8; its primary translation product, 22 kd, is processed to an extremely hydrophobic 4-kd protein that is associated with lipids in lamellar bodies.
SP-A is an innate host defense, large molecular, hydrophilic (water soluble) lectin coded on human chromosome 10 that regulates lung inflammation. SP-A contributes to the biophysical properties of surfactant primarily by decreasing protein-mediated inhibition of surfactant function. It binds to multiple organisms, such as group B streptococcus, Staphylococcus aureus, influenza virus, adenovirus, herpes simplex type 1, and respiratory syncytial virus. SP-A facilitates phagocytosis of pathogens by macrophages and their clearance from the airways. Mice that lack SP-A have no tubular myelin and have normal lung function and surfactant metabolism, indicating that SP-A is not a critical regulator of surfactant metabolism. Patients with SP-A deficiency have not been described.
SP-D is also a hydrophilic protein of 43 kd that is a collectin with structural similarities to SP-A. It has a collagenlike domain and a glycosylated region that gives it its lectinlike functions. SP-D is a large multimer that is synthesized by type II alveolar cells and Clara cells in addition to other epithelial cells in the body. It also binds pathogens and facilitates their clearance. The absence of SP-D results in increased surfactant lipid pools in the airspaces and emphysema in mice. No humans with SP-D deficiency have been described.
The components of pulmonary surfactant are synthesized in the Golgi apparatus of the endoplasmic reticulum of the type II alveolar cell. (See the image below.)
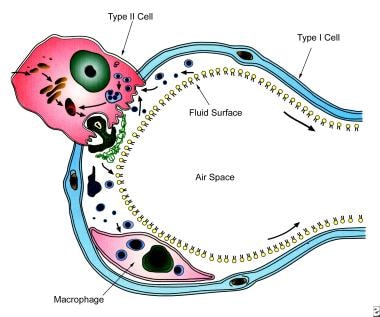 Schematic show surfactant metabolism, with a single alveolus is shown and the location and movement of surfactant components. Surfactant components are synthesized from precursors in the endoplasmic reticulum and transported through the Golgi apparatus by multivesicular bodies. Components are ultimately packaged in lamellar bodies, which are intracellular storage granules for surfactant before its secretion. After secretion (exocytosis) into the liquid lining of the alveolus, surfactant phospholipids are organized into a complex lattice called tubular myelin. Tubular myelin is believed to generate the phospholipid that provides material for a monolayer at the air-liquid interface in the alveolus, which lowers surface tension. Surfactant phospholipids and proteins are subsequently taken back into type II cells, in the form of small vesicles, apparently by a specific pathway that involves endosomes, and then are transported for storage into lamellar bodies for recycling. Alveolar macrophages also take up some surfactant in the liquid layer. A single transit of the phospholipid components of surfactant through the alveolar lumen normally requires a few hours. The phospholipid in the lumen is taken back into type II cell and is reused 10 times before being degraded. Surfactant proteins are synthesized in polyribosomes and extensively modified in the endoplasmic reticulum, Golgi apparatus, and multivesicular bodies. Surfactant proteins are detected in lamellar bodies or secretory vesicles closely associated with lamellar bodies before they are secreted into the alveolus.
Schematic show surfactant metabolism, with a single alveolus is shown and the location and movement of surfactant components. Surfactant components are synthesized from precursors in the endoplasmic reticulum and transported through the Golgi apparatus by multivesicular bodies. Components are ultimately packaged in lamellar bodies, which are intracellular storage granules for surfactant before its secretion. After secretion (exocytosis) into the liquid lining of the alveolus, surfactant phospholipids are organized into a complex lattice called tubular myelin. Tubular myelin is believed to generate the phospholipid that provides material for a monolayer at the air-liquid interface in the alveolus, which lowers surface tension. Surfactant phospholipids and proteins are subsequently taken back into type II cells, in the form of small vesicles, apparently by a specific pathway that involves endosomes, and then are transported for storage into lamellar bodies for recycling. Alveolar macrophages also take up some surfactant in the liquid layer. A single transit of the phospholipid components of surfactant through the alveolar lumen normally requires a few hours. The phospholipid in the lumen is taken back into type II cell and is reused 10 times before being degraded. Surfactant proteins are synthesized in polyribosomes and extensively modified in the endoplasmic reticulum, Golgi apparatus, and multivesicular bodies. Surfactant proteins are detected in lamellar bodies or secretory vesicles closely associated with lamellar bodies before they are secreted into the alveolus.
The components are packaged in multilamellar vesicles in the cytoplasm of the type II alveolar cell. They are secreted by a process of exocytosis, the daily rate of which may exceed the weight of the cell. Once secreted, the vesicles unwind to form bipolar monolayers of phospholipid molecules that depend on the apoproteins SP-B and SP-C to properly configure in the alveolus.
The lipid molecules are enriched in dipalmitoyl acyl groups attached to a glycerol backbone that pack tightly and generate low surface tension. Tubular myelin stores surfactant and depends on SP-B. Corners of the myelin lattice appear to be glued together with the large apoprotein SP-A, which may also have an important role in phagocytosis. Surfactant proteins are expressed in the fetal lung with increasing gestational age.
Patient education
Because the risk of prematurity and respiratory distress syndrome is increased for subsequent pregnancies, counsel the parents.
Education and counseling of parents, caregivers, and families of premature infants must be undertaken as part of discharge planning. These individuals should be advised of the potential problems infants with respiratory distress syndrome may encounter during and after their nursery stay. Audiovisual aids and handouts supplement such education.
Etiology
In premature infants, respiratory distress syndrome develops because of impaired surfactant synthesis and secretion leading to atelectasis, ventilation-perfusion (V/Q) inequality, and hypoventilation with resultant hypoxemia and hypercarbia. Blood gases show respiratory and metabolic acidosis that cause pulmonary vasoconstriction, resulting in impaired endothelial and epithelial integrity with leakage of proteinaceous exudate and formation of hyaline membranes (hence the name).
The relative deficiency of surfactant decreases lung compliance (see the image below) and functional residual capacity, with increased dead space. The resulting large V/Q mismatch and right-to-left shunt may involve as much as 80% of the cardiac output.
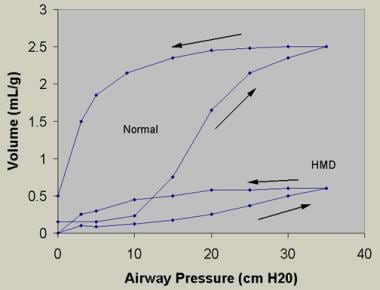 Bottom curve reflects findings from lungs obtained at postmortem from an infant with hyaline membrane disease (HMD). Lungs with HMD require far more pressure than to achieve a given volume of inflation than do lungs obtained from an infant dying of a nonrespiratory cause. Arrows indicate inspiratory and expiratory limbs of the pressure-volume curves. Note the decreased lung compliance and increased critical opening and closing pressures, respectively, in the premature infant with HMD.
Bottom curve reflects findings from lungs obtained at postmortem from an infant with hyaline membrane disease (HMD). Lungs with HMD require far more pressure than to achieve a given volume of inflation than do lungs obtained from an infant dying of a nonrespiratory cause. Arrows indicate inspiratory and expiratory limbs of the pressure-volume curves. Note the decreased lung compliance and increased critical opening and closing pressures, respectively, in the premature infant with HMD.
Hypoxia, acidosis, hypothermia, and hypotension may impair surfactant production and/or secretion. In many neonates, oxygen toxicity with barotrauma and volutrauma in their structurally immature lungs causes an influx of inflammatory cell, which exacerbates the vascular injury, leading to bronchopulmonary dysplasia (BPD). Antioxidant deficiency and free-radical injury worsen the injury.
Upon macroscopic evaluation, the lungs of affected newborns appear airless and ruddy (ie, liverlike). Therefore, the lungs require an increased critical opening pressure to inflate. Diffuse atelectasis of distal airspaces along with distension of distal airways and perilymphatic areas are observed microscopically. Progressive atelectasis, barotrauma or volutrauma, and oxygen toxicity damage endothelial and epithelial cells lining these distal airways, resulting in exudation of fibrinous matrix derived from blood.
Hyaline membranes that line the alveoli (see the image below) may form within a half hour after birth. In larger premature infants, the epithelium begins to heal at 36-72 hours after birth, and endogenous surfactant synthesis begins. The recovery phase is characterized by regeneration of alveolar cells, including type II cells, with a resultant increase in surfactant activity. The healing process is complex.
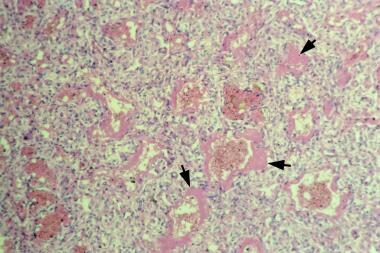 Microscopic appearance of lungs of an infant with respiratory distress syndrome. Hematoxylin and eosin stain shows hyaline membranes (pink areas).
Microscopic appearance of lungs of an infant with respiratory distress syndrome. Hematoxylin and eosin stain shows hyaline membranes (pink areas).
A chronic process often ensues in infants who are extremely immature and critically ill and in infants born to mothers with chorioamnionitis, resulting in BPD. In extremely premature infants, an arrest in lung development often occurs during the saccular stage, resulting in chronic lung disease termed "new" BPD.
Apoprotein deficiency
The hydrophobic SP-B and SP-C are essential for lung function and pulmonary homeostasis after birth. These proteins enhance the spreading, adsorption, and stability of surfactant lipids required to reduce surface tension in the alveolus. SP-B and SP-C participate in regulating intracellular and extracellular processes critical for maintaining respiratory structure and function. [1]
SP-B deficiency is an inherited deficiency caused by a pretranslational mechanism implied by the absence of messenger ribonucleic acid (mRNA). SP-B deficiency leads to death in term or near-term neonates and clinically manifests as respiratory distress syndrome with pulmonary hypertension, or congenital alveolar proteinosis. The genetic absence of SP-B is most often caused by a 2-base pair insertion (121 ins 2) that produces a frame shift and premature terminal signal, resulting in a complete absence of SP-B.
Approximately 15% of term infants who die of a syndrome similar to respiratory distress syndrome have SP-B deficiency. The lack of SP-B causes a lack of normal lamellar bodies in type II cells, a lack of SP-C, and the appearance of incompletely processed SP-C in the airspaces. These pro SP-C forms are diagnostic of SP-B deficiency.
Analysis of lung tissue with immunologic and biologic methods reveals an absence of one of the surfactant specific proteins, SP-B, and its mRNA. In an in-vitro study, critical structure and function in the N-terminal region of pulmonary SP-B was noted. W9 is critical to optimal surface activity, whereas prolines may promote a conformation that facilitates rapid insertion of the peptide into phospholipid monolayers compressed to the highest pressures during compression-expansion cycling.
Mutations of SP-B and SP-C cause acute respiratory distress syndrome and chronic lung disease that may be related to the intracellular accumulation of injurious proteins, extracellular deficiency of bioactive surfactant peptides, or both. Mutations in the gene for SP-C are a cause of familial and sporadic interstitial lung disease and emphysema as patients age. Mutations in other genes that cause protein misfolding and misrouting may contribute to the pathogenesis of chronic interstitial lung disease.
Hydrophilic SP-A and SP-D are lectins. In vivo and in vitro studies provide compelling support for SP-A and SP-D as mediators of various immune-cell functions. Studies have shown novel roles for these proteins in the clearance of apoptotic cells, direct killing of microorganisms, and initiation of parturition. None of the currently available surfactant preparations to treat respiratory distress syndrome have SP-A and SP-D.
ABCA3 mutations
Mutations in the adenosine triphosphate (ATP)–binding cassette gene (ABCA3) in newborns result in fatal surfactant deficiency. ABCA3 is critical for proper formation of lamellar bodies and surfactant function and may also be important for lung function in other pulmonary diseases. Because it is closely related to the ABCA1 - and ABCA4 -encoded proteins that transport phospholipids in macrophages and photoreceptor cells, it may have a role in surfactant phospholipid metabolism. [2]
The incidence of genetic abnormalities of pulmonary surfactant disorders is unknown. In a review of 300 term infants presenting as severe respiratory distress syndrome, 14% had SP-B deficiency and 14% had a deficiency of ABCA3.
Risk factors
The greatest risk factor for respiratory distress syndrome is prematurity, although the syndrome does not occur in all premature newborns. Other risk factors include maternal diabetes, cesarean delivery, and asphyxia. [3, 4]
Epidemiology
United States data
In the United States, respiratory distress syndrome has been estimated to occur in 20,000-30,000 newborn infants each year and is a complication in about 1% pregnancies. Approximately 50% of the neonates born at 26-28 weeks' gestation develop respiratory distress syndrome, whereas less than 30% of premature neonates born at 30-31 weeks' gestation develop the condition.
In one report, the incidence rate of respiratory distress syndrome was 42% in infants weighing 501-1500g, with 71% reported in infants weighing 501-750g, 54% reported in infants weighing 751-1000g, 36% reported in infants weighing 1001-1250g, and 22% reported in infants weighing 1251-1500g, among the 12 university hospitals participating in the National Institute of Child Health and Human Development (NICHD) Neonatal Research Network. [5]
International data
Respiratory distress syndrome is encountered less frequently in developing countries than elsewhere, primarily because most premature infants who are small for their gestation are stressed in utero because of malnutrition or pregnancy-induced hypertension. In addition, because most deliveries in developing countries occur at home, accurate records in these regions are unavailable to determine the frequency of respiratory distress syndrome.
Race-related demographics
Respiratory distress syndrome has been reported in all races worldwide, occurring most often in white premature infants.
Prognosis
Acute complications of respiratory distress syndrome include the following [6] :
-
Alveolar rupture
-
Infection
-
Intracranial hemorrhage and periventricular leukomalacia
-
Patent ductus arteriosus (PDA) with increasing left-to-right shunt
-
Pulmonary hemorrhage
-
Necrotizing enterocolitis (NEC) and/or gastrointestinal (GI) perforation
-
Apnea of prematurity
Chronic complications of respiratory distress syndrome include the following:
-
Bronchopulmonary dysplasia (BPD)
-
Retinopathy of prematurity (ROP)
-
Neurologic impairment
Alveolar rupture
Suspect an air leak (eg, pneumomediastinum, pneumopericardium, interstitial emphysema, pneumothorax) when an infant with respiratory distress syndrome suddenly deteriorates with hypotension, apnea, or bradycardia or when metabolic acidosis is persistent.
Infection
Infections may complicate the management of respiratory distress syndrome and may manifest in various ways, including failure to improve, sudden deterioration, or a change in white blood cell (WBC) count or thrombocytopenia. Also, invasive procedures (eg, venipuncture, catheter insertion, use of respiratory equipment) and use of postnatal steroids provide access for organisms that may invade the immunologically compromised host.
With the advent of surfactant therapy, small and ill infants are surviving, with an increased incidence of septicemia occurring in them secondary to staphylococcal epidermidis and/or candidal infection. When septicemia is suspected, obtain blood cultures from two sites and start appropriate antibiotics and/or antifungal therapy until culture results are obtained. Some neonatal ICUs use prophylactic fluconazole in the extremely premature infants, achieving a decrease in the incidence of candidal septicemia. [7]
Intracranial hemorrhage and periventricular leukomalacia
Intraventricular hemorrhage is observed in 20-40% of premature infants, with greater frequency in infants with respiratory distress syndrome who require mechanical ventilation. Cranial ultrasonography is performed in the first week in premature neonates younger than 32 weeks' gestation and at 36 weeks or at the time of discharge, or as indicated (eg, suspected seizures).
Use of antenatal steroids has decreased the frequency of intracranial hemorrhage in these patients with respiratory distress syndrome. Although a few studies have shown that prophylactic indomethacin therapy may decrease intraventricular hemorrhage in premature infants, its routine use is discouraged because of the risk of intestinal perforation. Hypocarbia and chorioamnionitis are associated with an increase in periventricular leukomalacia.
Patent ductus arteriosus with increasing left-to-right shunt
This shunt may complicate the course of respiratory distress syndrome, especially in infants weaned rapidly after surfactant therapy. Suspect patent ductus arteriosus (PDA) in any infant who deteriorates after initial improvement or who has bloody tracheal secretions.
Although helpful in the diagnosis of PDA, cardiac murmur and wide pulse pressure are not always apparent in critically ill infants. An echocardiogram enables the clinician to confirm the diagnosis. Infants requiring low fraction of inspired oxygen (FIO2) or who are clinically stable do not require treatment, as the PDA may close spontaneously. Ductal-dependent cardiac anomalies should be excluded prior to initiating therapy. Treat PDA with ibuprofen or indomethacin, which can be repeated during the first 2 weeks if the PDA reopens. [8] In refractory incidents of respiratory distress syndrome or in infants in whom medical therapy is contraindicated, surgically close the PDA.
Pulmonary hemorrhage
The occurrence of pulmonary hemorrhage increases in tiny premature infants, especially after surfactant therapy. Increase positive end-expiratory pressure (PEEP) on the ventilator and administer intratracheal epinephrine to manage pulmonary hemorrhage. In some patients, pulmonary hemorrhage may be associated with PDA; promptly treat pulmonary hemorrhage in such individuals.
In a retrospective study, intratracheal surfactant therapy was used successfully, with the rationale that blood inhibits pulmonary surfactant.
Necrotizing enterocolitis and/or GI perforation
Suspect NEC and/or GI perforation in any infant with abnormal abdominal findings on physical examination. Radiography of the abdomen assists in confirming their presence. Spontaneous perforation (not necessarily as part of NEC) occasionally occurs in critically ill premature infants and has been associated with the use of steroids and/or indomethacin.
Apnea of prematurity
Apnea of prematurity is common in immature infants, and its incidence has increased with surfactant therapy, possibly because of early extubation. Manage apnea of prematurity with methylxanthines (caffeine) and/or bubble or continuous flow nasal continuous positive airway pressure (CPAP), nasal intermittent ventilation, or with assisted ventilation in refractory incidents. Exclude septicemia, seizures, gastroesophageal reflux, and metabolic and other causes in infants with apnea of prematurity.
Bronchopulmonary dysplasia
BPD is a chronic lung disease defined as a requirement for oxygen at a corrected gestational age of 36 weeks. BPD is related directly to the high volume and/or pressures used for mechanical ventilation or to manage infections, inflammation, and vitamin A deficiency. BPD increases with decreasing gestational age.
Postnatal use of surfactant therapy, gentler ventilation, vitamin A, low-dose steroids, and inhaled nitric oxide may reduce the severity of BPD. [9]
Clinical studies have demonstrated various incidences of BPD, which has been attributed to increased survival of small and ill infants with respiratory distress syndrome. BPD may also be associated with gastroesophageal reflux or sudden infant death syndrome. Hence, consider these entities in infants with unexplained apnea before discharging them from the hospital.
Retinopathy of prematurity
Infants with respiratory distress syndrome who have a partial pressure of oxygen (PaO2) value of over 100mm Hg are at increased risk for ROP. Hence, closely monitor PaO2 and maintain it at 50-70mm Hg. Although pulse oximetry is used in all premature infants, it is not helpful in preventing ROP in tiny infants because of the flat portion of the oxygen-hemoglobin dissociation curve.
An ophthalmologist examines the eyes of all premature infants at 34 weeks' gestation and thereafter as indicated. If ROP progresses, laser therapy or cryotherapy is used to prevent retinal detachment and blindness. Closely monitor infants with ROP for refractive errors.
Intraocular bevacizumab, a monoclonal antibody targeting the vascular endothelial growth factor, has been used successfully to treat ROP. Although it is a promising therapy for ROP, further studies are needed before it can be recommended for routine use. [10]
Neurologic impairment
Neurologic impairment occurs in approximately 10-70% of infants and is related to the infant's gestational age, the extent and type of intracranial pathology, and the presence of hypoxia and infections. Hearing and visual handicaps may further compromise development in affected infants. Patients may develop a specific learning disability and aberrant behavior. Therefore, periodically follow up on these infants to detect those with neurologic impairment, and undertake appropriate interventions.
In a study that assessed the outcomes of 288 very preterm Chinese infants with severe respiratory distress syndrome on mechanical ventilation through 18 months of corrected age, the incidence of cerebral palsy and mental developmental index (MDI) less than 70 were highest among infants born at younger than 28 weeks' gestation compared to those born at 28-30 and 30-32 weeks' gestation. [11] Factors associated with cerebral palsy and an MDI below 70 were the administration of antenatal corticosteroids, decreased weight gain, and the presence of preeclampsia, fetal distress, and early/late bacteremia; factors that increased the risk of cerebral palsy and an MDI below 70 were increased length of mechanical ventilator support and blood transfusions. [11]
-
Chest radiographs in a premature infant with respiratory distress syndrome before and after surfactant treatment. Left: Initial radiograph shows poor lung expansion, air bronchogram, and reticular granular appearance. Right: Repeat chest radiograph obtained when the neonate is aged 3 hours and after surfactant therapy demonstrates marked improvement.
-
Microscopic appearance of lungs of an infant with respiratory distress syndrome. Hematoxylin and eosin stain shows hyaline membranes (pink areas).
-
Schematic outlines the pathology of respiratory distress syndrome (RDS). Infants may recover completely or develop chronic lung damage, resulting in bronchopulmonary dysplasia (BPD). FiO2 = fraction of inspired oxygen; HMD = hyaline membrane disease; V/Q = ventilation perfusion.
-
Bar chart demonstrates the composition of lung surfactant. About 1% of the 10% protein component comprises surfactant apoproteins; the remaining proteins are derived from alveolar exudate.
-
Schematic show surfactant metabolism, with a single alveolus is shown and the location and movement of surfactant components. Surfactant components are synthesized from precursors in the endoplasmic reticulum and transported through the Golgi apparatus by multivesicular bodies. Components are ultimately packaged in lamellar bodies, which are intracellular storage granules for surfactant before its secretion. After secretion (exocytosis) into the liquid lining of the alveolus, surfactant phospholipids are organized into a complex lattice called tubular myelin. Tubular myelin is believed to generate the phospholipid that provides material for a monolayer at the air-liquid interface in the alveolus, which lowers surface tension. Surfactant phospholipids and proteins are subsequently taken back into type II cells, in the form of small vesicles, apparently by a specific pathway that involves endosomes, and then are transported for storage into lamellar bodies for recycling. Alveolar macrophages also take up some surfactant in the liquid layer. A single transit of the phospholipid components of surfactant through the alveolar lumen normally requires a few hours. The phospholipid in the lumen is taken back into type II cell and is reused 10 times before being degraded. Surfactant proteins are synthesized in polyribosomes and extensively modified in the endoplasmic reticulum, Golgi apparatus, and multivesicular bodies. Surfactant proteins are detected in lamellar bodies or secretory vesicles closely associated with lamellar bodies before they are secreted into the alveolus.
-
Bottom curve reflects findings from lungs obtained at postmortem from an infant with hyaline membrane disease (HMD). Lungs with HMD require far more pressure than to achieve a given volume of inflation than do lungs obtained from an infant dying of a nonrespiratory cause. Arrows indicate inspiratory and expiratory limbs of the pressure-volume curves. Note the decreased lung compliance and increased critical opening and closing pressures, respectively, in the premature infant with HMD.
-
Effects of early treatment with low-dose inhaled nitric oxide (iNO) on brain injury (ie, grade 3-4 intracranial hemorrhage [ICH], periventricular leukomalacia [PVL], ventriculomegaly) in premature infants according to birth weight strata. iNO reduced ultrasonography findings of brain injury for the overall group (n = 793), with the largest effect in the 750-g to 999-g group (n = 280). Control = □; iNO = ■.
-
Effects of early treatment of preterm infants with low-dose inhaled nitric oxide (iNO) on bronchopulmonary dysplasia (BPD) incidence by birth weight strata. No difference in reduction was reported in infants weighing less than 1000 g (n = 129). Control = □; iNO = ■.
-
Effects of inhaled nitric oxide (iNO) survival without bronchopulmonary dysplasia (BPD) for infants aged 7-21 days. iNO increased survival without BPD in infants who were treated before age 14 days (n = 727).
-
Assisted ventilation newborn –Intubation and meconium aspiration. Video courtesy of Therese Canares, MD, and Jonathan Valente, MD, Rhode Island Hospital, Brown University.
Tables
- Table 1. Meta-Analysis of Early Versus Delayed Surfactant Treatment of RDS
- Table 2. Surfactant Preparations: Type, Source, Composition, Dosages, and Other Information
- Table 3. Results of a Meta-Analysis of Separate Clinical Trials of the Treatment of Respiratory Distress Syndrome With Natural or Synthetic Surfactant Preparations
- Table 4. Results of a Meta-Analysis of Separate Clinical Trials of the Prophylactic Use of Natural or Synthetic Surfactant Preparations
- Table 5. Results of a Meta-Analysis of Head-to-Head Trials With Natural Versus Synthetic Surfactants
- Table 6. Meta-Analysis of Clinical Trials Comparing Prophylactic Use of Surfactant Versus Rescue Treatment of Infants With Respiratory Distress Syndrome
- Table 7. Results of a Meta-Analysis of Clinical Trials to Compare Multiple Doses With a Single Dose of Surfactant
- Table 8. Inhaled Nitric Oxide Therapy in Preterm Infants and Outcome Measures
Outcome |
Number of Trials |
Relative Risk (95% CI) |
Relative Difference (95% CI) |
Pneumothorax |
3 |
0.70 (0.59, 0.82) |
-5.2% (-7.5%, -2.9%) |
Bronchopulmonary dysplasia (BPD) |
3 |
0.97 (0.88, 1.06) |
-1.2% (-4.6%, 2.2%) |
Mortality |
4 |
0.87 (0.77, 0.99) |
-2.8% (-5.5%, 0.0%) |
BPD or death |
3 |
0.94 (0.88, 1.00) |
-3.7% (-7.2%, 0.0%) |
Type |
Source |
Composition |
Dosing |
Comments |
Beractant (Survanta) |
Bovine lung mince |
Dipalmitoyl phosphatidylcholine (DPPC), tripalmitin, SP-B < 0.5%, SP-C 99% of TP wt/wt |
4mL/kg (100mg/kg), 1-4 doses every 6h |
Refrigerate |
Surfactant-TA (Surfacten) |
||||
Bovactant (Alveofact) |
Bovine lung lavage |
99% PL, 1% SP-B and SP-C |
45mg/mL |
From the Federal Republic of Germany |
Bovine lipid extract surfactant (bLES) |
Bovine lung lavage |
75% phosphatidylcholine (PC) and 1% SP-B and SP-C |
135mg/kg/dose (5mL/kg), 1-4 doses every 12h |
Canadian |
Infasurf |
Calf lung lavage |
DPPC, tripalmitin, SP-B 290g/mL, SP-C 360g/mL |
3mL/kg (105mg/kg), 1-4 doses every 6-12h |
6mL vials, refrigerate |
Calf lung surfactant extract (CLSE) |
Similar to Infasurf |
|||
Poractant alpha (Curosurf) |
Minced pig lung |
Phospholipids (DPPC, phosphatidylglycerol [PG]), neutral lipids, fatty acids; SP-B and SP-C; 80mg/mL of PL/mL [54mg PC (30.5mg DPPC and 1mg protein includes 0.3mg of SP-B)] |
Initially 2.5mL/kg (200mg/kg), followed by 1.25mL (100mg)/kg |
1.5 and 3mL |
Colfosceril palmitate (Exosurf) |
Synthetic |
85% DPPC, 9% hexadecanol, 6% tyloxapol |
5mL/kg (67.5mg/kg), 1-4 doses every 12h |
No longer available; lyophilized, dissolve in 8mL |
Lucinactant (Surfaxin) |
Synthetic |
Protein: KL4 (sinapultide) resembles SP-B; Phospholipids: DPPC, palmitoyloleoyl phosphatidylcholine (POPG) |
175 mg/kg/dose phospholipid |
FDA-approved March 2012; warmed for 15min at 44°C on a heating block, followed by vigorous shaking until a uniform, free-flowing suspension forms |
Artificial lung expanding compound (ALEC) |
Synthetic |
70% DPPC, 30% unsaturated phosphatidylglycerol |
No data |
Discontinued |
|
Natural Surfactant Treatment |
Synthetic Surfactant Treatment |
||
Outcome |
Number of Trials |
Relative Risk (95% Confidence Interval [CI]) Relative Difference (95% CI) |
Number of Trials |
Relative Risk (95% CI) Relative Difference (95% CI) |
Pneumothorax |
12 |
0.43 (0.35, 0.52) -17% (-21%, -13%) |
5 |
0.64 (0.55, 0.76) -9% (-12%, -6%) |
Bronchopulmonary dysplasia (BPD) |
9 |
0.94 (0.72, 1.22) -2% (-9%, 4%) |
5 |
0.75 (0.61, 0.92) -4% (-6%, -1%) |
Mortality |
12 |
0.68 (0.57, 0.80) -9% (-13%, -5%) |
6 |
0.73 (0.61, 0.88) -5% (-7%, -2%) |
BPD or death |
10 |
0.76 (0.65, 0.90) -14% (-21%, -7%) |
4 |
0.73 (0.65, 0.83) -8% (-11%, -5%) |
|
Natural Prophylaxis |
Synthetic Prophylaxis |
||
Outcome |
Number of Trials |
Relative Risk (95% CI) Relative Difference (95% CI) |
Number of Trials |
Relative Risk (95% CI) Relative Difference (95% CI) |
Pneumothorax |
8 |
0.35 (0.26, 0.49) -13% (-20%, -11%) |
6 |
0.67 (0.50, 0.90) -5% (-9%, -2%) |
BPD |
7 |
0.93 (0.80, 1.07) -4% (-9%, -3%) |
4 |
1.06 (0.83, 1.36) 1% (-4%, 6%) |
Mortality |
7 |
0.60 (0.44, 0.83) -7% (-12%, -3%) |
7 |
0.70 (0.58, 0.85) -7% (-11%, -3%) |
BPD or death |
7 |
0.84 (0.75, 0.93) -10% (-16%, -4%) |
4 |
0.80 (0.77, 1.03) -4% (-10%, 1%) |
Outcome |
Number of Trials |
Relative Risk (95% CI) |
Relative Difference (95% CI) |
Pneumothorax |
5 |
0.68 (0.56, 0.83) |
-4.1% (-6.3%, -2.0%) |
BPD |
4 |
0.97 (0.88, 1.07) |
-1.2% (-5.4%, -2.9%) |
Mortality |
7 |
0.88 (0.76, 1.02) |
-2.2% (-4.7%, 0.4%) |
BPD or death |
2 |
0.94 (0.87, 1.01) |
-3.6% (-8.0%, 0.8%) |
Outcome |
Number of Trials |
Relative Risk (95% CI) |
Relative Difference (95% CI) |
Pneumothorax |
6 |
0.62 (0.42, 0.89) |
-2.1% (-3.7%, -0.55) |
BPD |
7 |
0.95 (0.81, 1.11) |
-0.9% (-3.5%, 1.7%) |
Mortality |
6 |
0.59 (0.46, 0.76) |
-4.6% (-6.8%, -2.5%) |
BPD or death |
7 |
0.85 (0.76, 0.95) |
-4.5% (-7.4%, -1.5%) |
Infants < 30 wk of gestation |
|||
Mortality |
6 |
0.60 (0.47, 0.77) |
-6.5% (-9.6%, -3.4%) |
BPD or death |
7 |
0.86 (0.77, 0.96) |
-5.5% (-9.6%, -1.5%) |
Outcome |
Number of Trials |
Relative Risk (95% CI) |
Relative Difference (95% CI) |
Pneumothorax |
2 |
0.51 (0.30, 0.88) |
-8.7% (-15.4%, -2.0%) |
BPD |
1 |
1.10 (0.63, 1.93) |
1.2% (-5.8%, 8.3%) |
Mortality |
2 |
0.63 (0.57, 1.11) |
-7.0% (-14%, 0%) |
BPD or death |
1 |
0.80 (0.57, 1.11) |
-6.6%, (-16.2%. 3%) |
Study |
Number Enrolled |
Mean Gestational Age (wk) |
Mean Birth Weight (g) |
Mean Oxygen Index |
Placebo Death Rate |
Therapy Duration (d) |
Maximum Dose |
% Change in Death/BPD |
Kinsella et al [65] |
80 |
27 |
1000 |
30 |
53% |
7 |
5 ppm |
-15% |
Schreiber et al [66] |
201 |
27.2 |
970 |
10 |
22.5% |
7 |
10 ppm |
-15% |
Van Meurs et al [67] |
420 |
26 |
839 |
22 |
44% |
3 |
10 ppm |
-2% |
Ballard et al [68] |
587 |
26 |
760 |
7 |
6% |
24 |
20 ppm |
-11% |
Kinsella et al [69] |
793 |
25 |
792 |
5 |
25% |
14 |
5 ppm |
-4% |







