Background
In 1961, Childs et al published the earliest clinical report of a patient who was ultimately found to be affected by a deficiency of propionyl coenzyme A (CoA) carboxylase (ie, propionic acidemia). [1] These authors noted a series of severe ketoacidotic episodes in the child that were precipitated by protein ingestion (specifically, methionine and threonine administration) but manifested by marked elevations in plasma and urinary glycine levels. Because of these observations, the disease was given the name ketotic hyperglycinemia, a phenomenological term that inadvertently drew investigators' efforts toward a defect in glycine metabolism and delayed elucidation of the biochemical basis. The clinical hallmark of the disease is severe ketoacidosis of an episodic nature.
In 1969, Hsia et al described the underlying defect in propionate carboxylation that occurs in patients with ketotic hyperglycinemia. [2] Simultaneously, Morrow et al described the concurrence of methylmalonic acidemia and ketotic hyperglycinemia; thus, although the condition had been previously considered a single disorder, it was subsequently recognized on clinical grounds to be composed of least 2 different diseases. [3]
In 1971, subsequent studies by Hsia et al of the original patient's sister demonstrated a specific defect in propionyl CoA carboxylase. [4] The study also delineated propionic acidemia from methylmalonic acidemia as a distinct biochemical disorder. Subsequent work led to further delineation of another disorder, initially called multiple carboxylase deficiency, which includes deficiency of propionyl CoA carboxylase activity in addition to defects in other carboxylases.
The defect may be present at either of 2 different gene loci. One locus, on chromosome 13, controls synthesis of the α subunit of the tetrameric enzyme apoprotein; the second locus, on chromosome 3, controls synthesis of the β subunit. The 2 types of mutations are categorized as PCCA and PCCB (also PCCC) complementation groups, [5] distinguishable from each other by complementation studies of cultured fibroblasts in vitro. [6, 7]
Pathophysiology
The formation of propionyl CoA in human metabolism is derived from many sources, chiefly catabolism of a number of essential amino acids (isoleucine, valine, threonine, methionine). Other sources of propionyl CoA include odd chain-length fatty acids and the side chain of cholesterol, although these probably contribute very little in relation to the amino acid sources. Accumulation of the 3-carbon fatty acyl-CoA within the mitochondrion leads to decreased free CoA for other reactions, which is alleviated by conversion of propionyl CoA to propionyl-carnitine.
Propionyl-carnitine is transported out of the cell and excreted in urine; the mitochondrial CoA, thus freed, can participate in other reactions or once again become involved in formation of propionyl CoA. The relative reaction rates of these simultaneous processes stand between a tenuously balanced state of equilibrium and severe ketoacidosis. Carnitine deficiency can precipitate a clinical episode by disruption of the balance. Obviously, enhanced dietary protein intake has the same net effect by flooding the mitochondrion with propionyl CoA.
A common clinical finding is mild-to-moderate blood ammonia elevation, which may contribute by direct neurotoxicity to changes in a patient's mentation. Studies suggest that the underlying cause of the hyperammonemia is the inhibition of N -acetylglutamate synthase (NAGS) activity by free propionic acid. Since N -acetylglutamate (NAG) is the allosteric activator of carbamoylphosphate synthase, the entry step into the urea cycle, decreased ureagenesis occurs with accumulation of free ammonia. [8] See the image below.
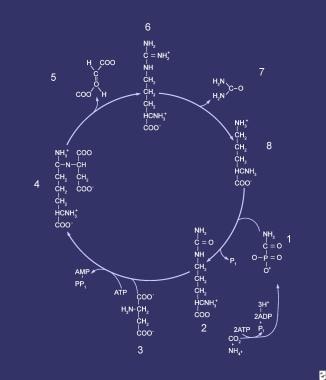 Urea cycle. Compounds that comprise the urea cycle are numbered sequentially, beginning with carbamyl phosphate. At the first step (1), the first waste nitrogen is incorporated into the cycle; also at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (4); the reaction is mediated by argininosuccinate (ASA) synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Urea cycle. Compounds that comprise the urea cycle are numbered sequentially, beginning with carbamyl phosphate. At the first step (1), the first waste nitrogen is incorporated into the cycle; also at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (4); the reaction is mediated by argininosuccinate (ASA) synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Additional evidence suggests that plasma glutamine-to-glutamate ratios decrease with increasing plasma ammonia concentration; simultaneously, urinary methylcitrate excretion increases while urinary citrate diminishes. Taken together, these data might suggest mitochondrial impairment, with inability to produce adequate alpha-ketoglutarate as substrate for formation of glutamate. This hypothesis, although attractive, has not been validated. The free organic acid has also been demonstrated to inhibit bone marrow production of leukocytes, red cells, and platelets. Pancytopenia is common and usually occurs 2-3 days after the acute presentation.
The reason for elevation of serum glycine levels is unclear, although studies show inhibition of glycine cleavage to 1-carbon fragments. Unlike ketoacidosis and hyperammonemia, elevation of glycine in the circulation is not known to be harmful. Thus, propionic acidemia manifests as clinical signs and symptoms of acidosis and hyperammonemia, including tachypnea, vomiting, lethargy, irritability, shock, coma, and death.
Epidemiology
Frequency
United States
The incidence rate is estimated to be approximately 1 per 100,000 live births. The caveat here is that population screening programs for the disease are not available in most areas; thus, this figure rests on clinical diagnosis. Because mild and even asymptomatic cases in which persons are genetically affected have been reported, the incidence is likely an under-representation of the true occurrence rate of homozygosity in the US population.
International
Although the disease is generally quite rare, the incidence rate has been reported to be as high as 1 per 2000 to 1 per 5000 births in areas of the world with restricted gene pools, such as Saudi Arabia.
Mortality/Morbidity
In most patients in whom presentation occurs in infancy with full-blown symptoms, the morbidity is very high. Severe ketoacidosis with pH values as low as 6.8 may cause circulatory shock, hypoxia, and irreparable brain damage. Repetitive hyperammonemia causes neurotoxicity with neuronal cell death leading to intellectual disability.
The pancytopenia commonly seen after clinical presentation renders the patient vulnerable to infection, which may become overwhelming and result in death. Death can occur at any time from an acute episode.
Cardiomyopathy (both hypertrophic and dilated) has become recognized as a frequent complication of propionic acidemia. [9, 10] Prolonged QTc interval, with or without evidence of cardiomyopathy, is also seen, and may be a cause of unexplained sudden death in these individuals. [11] One study suggests that the cardiomyopathy is reversible following orthotopic liver transplantation. [12]
Sex
Propionyl CoA carboxylase deficiency is inherited as an autosomal recessive trait; therefore, no sex bias is observed.
Age
Like many autosomal recessive traits, multiple mutations at a single gene locus can account for various clinical severities, especially given the potential for mixed heterozygosity.
Prognosis
Although less-severely affected patients have been reported, most individuals with propionic acidemia have a classic presentation and course and a guarded prognosis. Survival is in question, and significant brain damage is likely.
Patient Education
Parents must be taught to strictly adhere to the dietary regimen as prescribed. They also must be made aware of the importance of follow-up for adjustment of diet to meet the requirements for growth.
Patients must be seen as early as feasible during the course of any intercurrent illness. Treat patients with intravenous glucose and bicarbonate immediately if indicated.
-
Urea cycle. Compounds that comprise the urea cycle are numbered sequentially, beginning with carbamyl phosphate. At the first step (1), the first waste nitrogen is incorporated into the cycle; also at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (4); the reaction is mediated by argininosuccinate (ASA) synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.









