Practice Essentials
Osteogenesis imperfecta (OI) is a disorder of bone fragility chiefly caused by mutations in the COL1A1 and COL1A2 genes, which encode type I procollagen. [1, 2, 3] Four types of osteogenesis imperfecta were originally described by Sillence in 1979 and are now used broadly as the Sillence Criteria. [4] The Nosology and Classification of Genetic Skeletal Disorders provides similar categorization in the 2010 revision. [5] Precise typing is often difficult, and depends in large degree on the experience of the clinician. Severity ranges from mild forms to lethal forms in the perinatal period. Additional genes have been discovered in which mutations can also cause brittle bones. These are typically clinically indistinguishable and are considered by most to be subtypes of osteogenesis imperfecta. [6] Examples of common radiologic findings of osteogenesis imperfecta are shown in the images below.
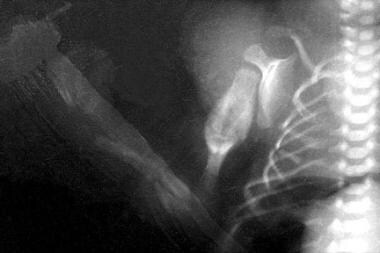 Acute fractures are observed in the radius and ulna. Multiple fractures can be seen in the ribs. Old healing humeral fracture with callus formation is observed.
Acute fractures are observed in the radius and ulna. Multiple fractures can be seen in the ribs. Old healing humeral fracture with callus formation is observed.
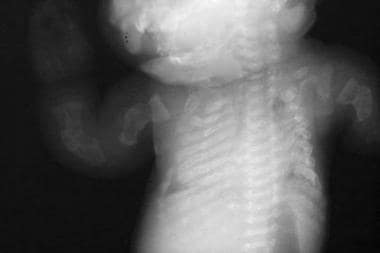 Beaded ribs. Multiple fractures are seen in the long bones of the upper extremities.
Beaded ribs. Multiple fractures are seen in the long bones of the upper extremities.
Physical characteristics of osteogenesis imperfecta
Individuals with type III (severe, progressively deforming) OI may have joint hyperlaxity; muscle weakness; chronic, unremitting bone pain; and skull deformities (eg, posterior flattening) due to bone fragility during infancy. Limb shortening and progressive deformities can occur, and patients have a triangular face with frontal and temporal bossing; malocclusion is common. The sclera have variable hues.
Workup in osteogenesis imperfecta
Results from routine laboratory studies in patients with OI are usually within reference ranges, and they are useful in ruling out other metabolic bone diseases, such as hypophosphatasia or inherited forms of rickets.
Collagen synthesis analysis has historically been performed by culturing dermal fibroblasts obtained during skin biopsy. This testing modality has fallen out of favor, given that molecular diagnostic techniques are more accurate, have increased in availability, and have decreased in cost. Widespread adoption of next-generation sequencing (NGS) technology has allowed for parallel sequencing of many genes. Thus, there is little or no difference in cost or turnaround time for multigene panels compared with limiting investigation to the two procollagen I genes.
Obtain a radiographic skeletal survey after birth. Prenatal ultrasonography can be used to detect limb-length abnormalities at 15-18 weeks’ gestation.
The width of biopsy cores, the width of the cortex, and the volume of cancellous bone are decreased in all types of osteogenesis imperfecta. The number and thickness of trabeculae are reduced.
Management
Cyclic administration of intravenous pamidronate reduces the incidence of fracture and increases bone mineral density, while reducing pain and increasing energy levels. [7] Intravenous zoledronate has been increasingly used in children with OI, given its shorter infusion times and longer infusion intervals. Observational studies have shown similar effects to that of pamidronate, with increased bone mineral density, decreased fracture rate, and improvement in pain. [8] There is scant evidence supporting the use of bisphosphonates in adults with OI, although they continue to be widely used; anecdotally, some patients experience a subjective decrease in pain. [9] The bisphosphonate risedronate may have some effect in reducing fractures in patients with OI. [10]
Nutritional evaluation and intervention are paramount to ensure appropriate intake of calcium and vitamin D. Caloric management is important, particularly in adolescents and adults with severe forms of OI.
Orthopedic surgery is one of the pillars of treatment for patients with OI. [11] Surgical interventions include intramedullary rod placement, surgery to manage basilar impression, and correction of scoliosis.
Pathophysiology
The most widely used classification of osteogenesis imperfecta, published by Sillence et al in 1979, does not include additional forms of the disorder, which were discovered as a result of improvements in molecular diagnostics. [12] Rather, these forms of osteogenesis imperfecta are caused by genes that interact with collagen I or in the complex relationship between formation and remodeling of bone. They are molecularly distinct from osteogenesis imperfecta caused by COL1A1 or COL1A2 but form an example of locus heterogeneity. Forlino and Marini, in 2015, offered an alternate way of understanding the genetics of osteogenesis imperfecta, using five functional categories, as follows [13] :
-
Group A: Primary defects in collagen structure or function ( COL1A1, COL1A2, BMP1)
-
Group B: Collagen modification defects ( CRTAP, LEPRE1, PPIB, TMEM38B)
-
Group C: Collagen folding and cross-linking defects ( SERPINH1, FKBP10, PLOD2)
-
Group D: Ossification or mineralization defects ( IFITM5, SERPINF1)
-
Group E: Osteoblast development defects with collagen insufficiency ( WNT1, CREB3L1, SP7)
This classification system has not been integrated into widespread use but offers significant streamlining of categories into intellectually satisfying divisions.
COL1A1/COL1A2 (Types I-IV)
Type I collagen fibers are found in the bones, organ capsules, fascia, cornea, sclera, tendons, meninges, and dermis. Type I collagen, which constitutes approximately 30% of the human body by weight, (90% of the protein within bone) is defective in osteogenesis imperfecta. In structural terms, type I collagen fibers are composed of a left-handed helix formed by intertwining of pro-alpha 1 and pro-alpha 2 chains. Mutations in the genes that encode these chains cause osteogenesis imperfecta (COL1A1 at 17q21 and COL1A2 at 7q22.1, respectively).
Qualitative defects (eg, an abnormal collagen I molecule) and quantitative defects (eg, decreased production of normal collagen I molecules) are described. Quantitative defects most frequently result from missense mutations in a glycine-coding codon and cause disease due to a dominant negative effect. There is not a perfect correlation between genotype and phenotype, but a few observations have been made. First, generally speaking mutations in the COL1A1 gene will tend to be more severe than a corresponding mutation in the COL1A2 gene, likely due to the fact that that two alpha 1 chains are required and only one alpha 2 chain is required to make the procollagen I heterotrimer. Second, mutations located at the carboxyl terminal domain of each of the genes are more severe than in other domains. In particular, missense mutations in COL1A1 in this region are frequently (but not always) lethal. Third, size and polarity matter to the severity of the mutation. As an example, a substitution of glycine for alanine, which is only slightly larger, nonpolar, and with similar chemical properties, is likely to result in a milder phenotype. Contrast this with a substitution of glycine for glutamic acid at the same position, which is large, negatively charged, and with different chemical properties and is more likely to be severe or even lethal. Quantitative defects often result from nonsense mutations but can result from larger deletions in one of the procollagen genes and cause disease due to haploinsufficiency effect, as the prematurely truncated mRNA undergoes nonsense-mediated decay (NMD); while the patient is left with structurally normal collagen, he or she makes only half the normal amount. These patients have mild nondeforming (Type I) osteogenesis imperfecta.
A study by Balasubramanian et al indicated that COL1A1/COL1A2 mutations consistently result in collagen fibril diameter variability and collagen flowers. [14]
Osteogenesis Imperfecta with Calcification of the Interosseous Membranes (Type V)
Patients with this form of osteogenesis imperfecta generally have moderate severity disease but frequently develop hyperplastic calluses in long bones after having a fracture or orthopedic surgery that involves osteotomies. The size and shape of the callus may remain stable for many years after a rapid growth period, but in some cases slowly involutes. Patients also frequently develop calcification of the forearm interosseous membrane and dislocation of the radial head, which results in difficulties with supination and pronation. They may also have subphyseal metaphyseal radiodensity seen on radiography. [15] This condition is caused by heterozygous mutations in the IFITM5 gene, and inheritance is autosomal dominant. [16]
Histology of bone showed that the lamellae are arranged in an irregular fashion and in some cases appeared meshlike, as opposed to the typical parallel arrangement in patients with osteogenesis imperfecta.
Other Forms of Osteogenesis Imperfecta
SERPINFI (Type VI)
Patients with this form of osteogenesis imperfecta generally have moderate severity disease. It was observed that many patients with this form of osteogenesis imperfecta do not have fractures at birth and only develop them later in infancy or as toddlers. Patients with this form of osteogenesis imperfecta have been described as having variably blue or white sclerae and have normal teeth. [17] This condition is caused by homozygous mutation in the SERPINF1 gene, and inheritance is autosomal recessive. [18]
Histology of bone showed a characteristic fish-scale appearance to the lamellae and at least one author has commented on the presence of large quantities of unmineralized osteoid on biopsy specimens.
CRTAP/LEPRE1/PPIB (Types VII-IX)
Patients with osteogenesis imperfecta caused by mutations in these three genes generally have severe disease, with examples of lethal disease being noted. Cartilage-associated protein (CRTAP) is a protein required for prolyl 3-hydroxylation, and with the protein products of the LEPRE1 and PPIB genes, forms a heterotrimeric protein that is crucial for proper posttranslational modification of collagen I. Osteogenesis imperfecta caused by mutations in CRTAP have been designated type VII disease, whereas osteogenesis imperfecta caused by mutations in LEPRE1 and PPIB are designated type VIII and type IX disease, respectively. [19] These conditions are caused by homozygous or compound heterozygous mutations and are inherited in an autosomal recessive manner.
SERPINH1 (Type X)
Report of only one patient with this form of osteogenesis imperfecta could be found in the literature. This male patient was born to a consanguineous union of Saudi Arabian origin, and his parents were reported as clinically normal. He was reported to have severe deforming osteogenesis imperfecta with dentinogenesis imperfecta, nephrocalcinosis, and chronic lung disease. He died at age 3 years from respiratory complications. Genetic testing of this child found a previously described homozygous mutation in the SERPINH1 gene. [20] The protein product of SERPINH1 is a chaperone in the endoplasmic reticulum (ER) that appears to function in collagen trafficking.
FKBP10 (Type XI)
A small number of patients with this form of osteogenesis imperfecta have been reported and severity has been moderate to severe. It is caused by homozygous mutations in the FKBP10 gene and is inherited in an autosomal recessive manner. [21] It is understood that some patients with Bruck syndrome, type II also have mutations in this gene, but the full spectrum of clinical findings is not understood, and it has been proposed to designate FKBP10 -related disorders as recessive forms of progressive deforming osteogenesis imperfecta with or without joint contractures. [22] The protein product of FKBP10 is a chaperone that participates in collagen folding and mutation impairs its secretion. [23]
Histology of bone showed a distorted lamellar structure and a fish scale pattern.
SP7 (Type XII)
Only one patient with this form of osteogenesis imperfecta has been reported and had moderate severity disease. This male patient was the product of a second-cousin union and in addition to fractures had delay in tooth eruption, normal hearing, and white sclerae. A sibling was similarly affected, but also died of a presumably unrelated congenital heart defect. The surviving child had homozygous deletions in the SP7 gene, and this form of osteogenesis imperfecta is inherited in an autosomal recessive fashion. [24] The protein product of SP7 is required for osteoblast differentiation and bone formation. In a null mutant animal model, no cortical bone or bone trabeculae were formed through intramembraneous or endochondral ossification. [25]
BMP1 (Type XIII)
A small number of patients with this form of osteogenesis imperfecta have been described and have generally had severe disease with frequent fractures and severe growth deficiency. However, two siblings described had only borderline low bone density. Patients with this form of osteogenesis imperfecta were described with normal teeth and blue sclerae. [26] This form of osteogenesis imperfecta is caused by homozygous mutation in the BMP1 gene, and it is inherited in an autosomal recessive manner. The BMP1 protein product appears to play important roles in osteogenesis, but also is thought to have a role in the removal of C-propeptides from certain procollagens, including type I procollagen. [27]
TMEM38B (Type XIV)
This form of osteogenesis imperfecta has been described in three consanguineous families of Saudi origin, and three different consanguineous families of Israeli Bedouin origin. Affected individuals had osteogenesis imperfecta of variable severity without blue sclerae, dentinogenesis imperfecta, or hearing loss. [28] This form of OI is caused by homozygous mutation in the TMEM38B gene and is inherited in an autosomal recessive manner. [29]
WNT1 (Type XV)
This recently discovered form of osteogenesis imperfecta has now been described in a number of families and causes moderate to severe disease. Patients with this condition have had short stature, blue or white sclerae, and normal hearing. A subset of patients have been seen with brain malformations and developmental delay. This form of OI is caused by homozygous or compound heterozygous mutations in the WNT1 gene and is inherited in an autosomal recessive manner. [30] Canonical WNT/beta-catenin signaling has been shown to be crucial for the differentiation of osteoblasts and further bone development. [31]
CREB3L1 (Type XVI)
CREB3L1 encodes the endoplasmic reticulum stress transducer protein OASIS, which regulates the expression of type 1 procollagen. Biallelic mutation in this gene can be expected to produce insufficiency of type I procollagen. Two siblings have been reported with this form of OI, and both were homozygous for the same deletion. This deletion is some 91 kB in size and renders patients nullisomic for the CREB3L1 locus. The first affected patient was male and was known to be affected by severe OI. This patient was small for gestational age, with multiple fractures in utero. Postnatally, he developed a number of fractures and was described with beaded ribs. He also had several bouts of pneumonia and died at 9 age months, presumably from respiratory complications, although this was not explicitly stated. The second affected sibling was also male and termination was performed at 19 weeks of gestation. The parents and a healthy older sister did not have similar findings of bone fragility but were described with blue sclerae. The mother had velvety skin and small-joint hypermobility. The father also had velvety skin, as well as conductive hearing loss. [32]
SPARC (Type XVII)
SPARC (secreted protein, acidic, cysteine-rich) is a glycoprotein that binds to multiple matrix proteins, including collagen I. Two unrelated patients have been described as homozygous for pathogenic variants in the SPARC gene; both had significant bone fragility and had been previously diagnosed with Type IV OI based on phenotype alone. The first patient was of North African origin and in addition to her fractures, experienced a neonatal intraventricular hemorrhage. At the time of her evaluation by the authors, she had sustained 10 long-bone fractures but was of normal stature. She had joint hypermobility and humeral bowing, and her bone mineral density was low for her age. The second patient was born of a consanguineous union and was of Indian descent. She presented initially with hip dislocation and was subsequently observed to have hypotonia and motor delay. She also had normal stature, as well as joint laxity and soft skin. Her bone mineral density was also low for her age. Molecular diagnosis was obtained in these patients using whole exome sequencing (WES). The first patient was homozygous for the pathogenic variant c.497G>A (p.Arg166His), and the second patient was homozygous for the pathogenic variant c.787G>A (p.Glu263Lys). It was observed that these pathogenic variants were both in highly conserved regions of the protein. Further investigation revealed that each was located in different portions of the extracellular collagen binding portion of the SPARC protein. [33]
TENT5A (FAM46A) (Type XVIII)
TENT5A (terminal nucleotidyltransferase 5A), also called FAM46A, is an example of a class of enzymes that catalyze the transfer of a nucleoside monophosphate molecule from nucleoside triphosphate to a hydroxyl group in a different molecule. Changes in the TENT5A enzyme cause a recessive form of OI, and a small number of patients have been reported with homozygous pathogenic variants in the TENT5A gene. Doyard et al reported a small case series of four patients in three families with this form of OI. All affected patients were known or suspected to be consanguineous. Patients in this series had variable presentation, but consistent features included typical OI traits such as bowing of the limbs, frequent fractures, wormian bones, blue sclerae, and joint hypermobility. One patient died suddenly at age 4 years for unknown reasons that presumably were unrelated to the OI diagnosis. [34]
MBTPS2 (Type XIX)
MBTPS2 (membrane-bound transcription factor protease, site 2) is involved in transcription control and in the ER stress response. Type XIX OI is an X-linked recessive form of the disease that has been seen in several males in two unrelated families, one from Thailand and one from Germany. Patients presented with multiple fractures, bowing of long bones, low bone mineral density, pectus excavatum, and moderate to severe short stature. There is no evidence of a phenotype for carrier females. [35]
MESD (Type XX)
MESD (mesoderm development LRP chaperone) is involved in mesodermal development and is believed to be involved in embryonic polarity and mesodermal induction. More importantly in this context, its protein functions as a chaperone for LRP5 and LRP6, which are coreceptors in the Wnt pathway. [36] . In 2019, Moosa and colleagues reported on five patients from several consanguineous families. Presentations were severe, and patients had a notable trend toward respiratory involvement, with two of the five patients dying of respiratory failure prior to age 2 years. Fractures occurred early in life and involved long bones and vertebrae. Dentinogenesis imperfecta was not seen, but oligodontia was common. [37]
KDELR2 (Type XXI)
KDELR2 (KDEL endoplasmic reticulum protein retention receptor 2) is involved in ER trafficking. It is believed that alterations in KDELR2 cause an OI phenotype due to the inability of the mutant KDELR2 to bind to the HSP47 gene product; this alters the ability of HSP47 to dissociate from collagen I, thereby disrupting fiber formation. [38, 39] Patients with this form of OI present with short stature, blue or white sclerae, typical chest deformity, and variability in fracture incidence similar to other forms of OI. Both limb bowing and dentinogenesis imperfecta have been seen in some, but not all, patients. Speech and motor delays have also been reported in some patients, but when intelligence was reported, it was normal. [39, 40]
Osteogenesis imperfecta with congenital joint contractures, Types 1 and 2 (Bruck syndrome)
Patients with Bruck syndrome have congenital brittle bones prone to fracture, as well as congenital joint contractures and pterygia. They also have short stature, severe limb deformity, wormian bones, and progressive scoliosis which can be severe. Patients have generally been described with normal hearing, no dentinogenesis imperfecta, and white sclerae. Two forms of Bruck syndrome have been delineated with molecular testing, but appear to be clinically indistinguishable. Bruck syndrome 1 is caused by homozygous mutation in the FKBP10 gene [41] , while Bruck syndrome 2 is caused by homozygous mutation in the PLOD2 gene. [42] Both disorders are inherited in an autosomal recessive manner. It has been suggested that the defect underlying Bruck syndrome is a deficiency of bone-specific telopeptide lysyl hydroxylase, which results in aberrant bone collagen crosslinking. [43]
Cardiovascular concerns in osteogenesis imperfecta
A number of published reports have identified patients with OI who have had aortic and other vascular dissections, and there are reasonable concerns about increased risk in this population. A study by Rush et al evaluated 100 children and adolescents with OI and found that patients with Types III and IV had significantly larger aortic diameters compared with size-matched controls. The investigators also determined that patients with Type I OI had normal aortic diameters but larger and thinner left ventricles compared with size-matched controls. No patient in this study had cardiovascular findings that were immediately actionable, but since reports of potentially lethal cardiovascular complications have been described in adult patients, a screening echocardiogram by age 18 in patients with OI is recommended. [44] No truly evidence-based guidelines for surveillance of cardiovascular disease in OI have been published.
Epidemiology
Frequency
United States
The prevalence of OI is estimated to be 1 per 15,000 live births; [45] however, the mild form is underdiagnosed, and the actual prevalence may be higher.
International
Prevalence appears to be similar worldwide, although there may be an increased risk of recessive forms of OI in populations with a high degree of consanguinity.
A retrospective study from Spain, by Darbà and Marsà, found the hospital incidence of OI to be 5.64 per 100,000 patients between the years 2000 and 2017, and the incidence at birth to be 10.14 per 100,000 children. The incidence is similar to that which was previously reported, but the incidence at birth of 1 in 9800 is higher than past estimates. It bears mention that the reported experience in Spain may not be generalizable globally. [46]
Race
No differences based on race are reported.
Sex
No differences based on sex are reported.
Age
The age when symptoms (ie, fractures) begin widely varies. Patients with mild forms of OI generally do not present at birth, instead often presenting with fracture after incidental trauma. Patients with severe cases typically present with fractures in utero.
-
Acute fractures are observed in the radius and ulna. Multiple fractures can be seen in the ribs. Old healing humeral fracture with callus formation is observed.
-
Beaded ribs. Multiple fractures are seen in the long bones of the upper extremities.
-
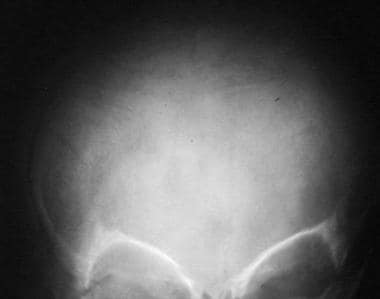
Wormian bones are present in the skull.
-
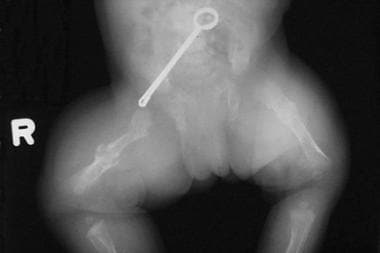
This newborn has bilateral femoral fractures.








