Practice Essentials
Immotile cilia syndrome (ICS) is an autosomal recessive disease with extensive genetic heterogeneity characterized by abnormal ciliary motion and impaired mucociliary clearance. Ultrastructural and functional defects of cilia result in the lack of effective ciliary motility, causing abnormal mucociliary clearance. This leads to recurrent or persistent respiratory infections, sinusitis, otitis media, and male infertility. In 50% of the patients, ICS is associated with situs inversus.
Signs and symptoms
The following signs and symptoms may be present in patients with primary ciliary dyskinesia:
-
Nasal mucosal congestion
-
Mucopurulent nasal discharge
-
Nasal obstruction
-
Mouth breathing and halitosis
-
Nasal polyps
-
Inflammation of tympanic membranes
-
Perforation with purulent discharge
-
Hearing loss
-
Respiratory distress
-
Retractions
-
Hypoxia
-
Crackles, wheeze
-
Apex beat and heart sounds on the right side, if associated dextrocardia is present
-
Evidence of situs inversus, such as the spleen and liver on the incorrect side
-
Digital clubbing in cases with chronic and recurrent lower respiratory infections
See Presentation for more detail.
Diagnosis
Laboratory studies
-
Genetic testing
-
Ciliary biopsy for electron microscopy
Imaging studies
-
Chest roentgenography
-
Direct video cinematography or oscillography
-
Digital high-speed video imaging
See Workup for more detail.
Management
There are no specific therapies, or prospective, randomized clinical trials on monitoring or treating primary ciliary dyskinesia. General principles of airway clearance and antibiotic therapy used for cystic fibrosis (CF) or non-CF bronchiectasis should be followed. Surgery may be indicated when antibiotic therapy has not been helpful.
See Treatment and Medication for more detail.
Patient education
Genetic counseling should be offered to parents of newly diagnosed infants and children. The importance of regular health monitoring should be emphasized. Counsel patients to avoid smoke, allergens, environmental irritants, and exposure to respiratory pathogens.
Background
In 1933, Kartagener described a unique syndrome characterized by the triad of situs inversus, chronic sinusitis, and bronchiectasis, which was dubbed Kartagener syndrome. [1, 2] Later, patients with this condition were noted to have defects in the ultrastructure of cilia. Afzelius coined the term immotile cilia. [3] Later studies showed that disorganized motion, rather than immotile cilia, resulted in the uncoordinated and ineffective ciliary beat, hence the term ciliary dyskinesia syndrome (CDS). Because transient ciliary dyskinesia may be acquired following epithelial injury from viral respiratory tract infections or exposure to pollutants, [4, 5] the term primary ciliary dyskinesia (PCD) is used to describe the genetic defect and to differentiate it from acquired defects.
Dysfunction of the axonemal structure has been linked to the emerging class of disorders collectively known as ciliopathies, which includes PCD/Kartagener syndrome, Bardet-Biedl syndrome, hydrocephalus, polycystic kidney disease, polycystic liver disease, nephrolithiasis, Meckel-Gruber syndrome, and Joubert syndrome. [6] A report of 9 patients with PCD from an inbred Amish community reported genetic heterogeneity. [7]
Review of normal and abnormal ciliary ultrastructure
The epithelial lining of the large airways and contiguous structures, including the paranasal sinuses, middle ears, and posterior nose, consists of ciliated pseudostratified columnar epithelium. Ciliated cells are also found in the ependymal lining of the brain and fallopian tubes. In addition, the spermatozoal flagella (tail of spermatozoa) has a core structure that is identical to cilia.
Each matured ciliated cell has up to 200 cilia. Each cilium has an array of longitudinal microtubules arranged as 9 doublets formed in an outer circle around a central pair (see image below). The main structural protein of these doublets is tubulin. The microtubules are anchored by a basal body in the apical cytoplasm of the cell. Radial spokes connect the outer microtubular doublets with a central sheath of protein around the central tubules.
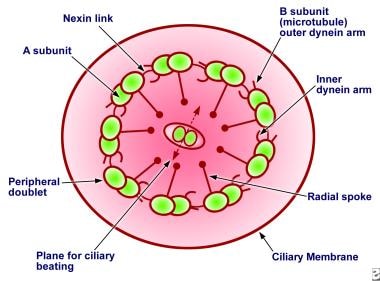 Diagram showing the cross-section of normal cilia showing its ultrastructure. Important components are labeled.
Diagram showing the cross-section of normal cilia showing its ultrastructure. Important components are labeled.
Cross-section of the cilia (see image below) reveals inner and outer dynein arms, which are attached to the A subunit of each microtubule doublet. The inner dynein arms are longer and form a hook, whereas the outer dynein arms are short and straight. Dynein, a type of ATPase, provides energy for microtubule sliding and the longitudinal displacement of adjacent microtubular doublets, resulting in ciliary bending. The protein nexin links the outer microtubular doublets, creating a circumferential network as straplike bands. Because nexin links maintain axonemal relationships while the basal bodies anchor the microtubules, the sliding of the outer microtubule results in bending of the cilium.
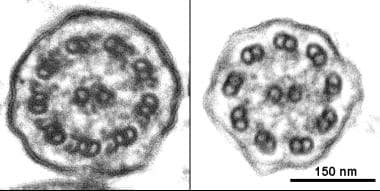 Ciliary ultrastructure, Left, Normal cilium from a healthy individual in which both inner and outer dynein arms can clearly identified. Right, the absence of outer and inner dynein arms in a patient with primary ciliary dyskinesia. Image courtesy of J. Carson, PhD, University of North Carolina.
Ciliary ultrastructure, Left, Normal cilium from a healthy individual in which both inner and outer dynein arms can clearly identified. Right, the absence of outer and inner dynein arms in a patient with primary ciliary dyskinesia. Image courtesy of J. Carson, PhD, University of North Carolina.
Ciliary movement involves 2 phases: an effective stroke phase that sweeps forward and a recovery phase during which the cilia bend backward and extend into the starting position for the stroke phase. The mucous lining present on the respiratory epithelium has an inner serous layer called the sol phase, in which the cilia recover from their active beat, and an outer, more viscous layer, the gel phase. The tips of the cilia contact the gel layer during the stroke phase to propel the secretions forward, but the cilia lose contact with the gel layer of the mucus during the recovery phase.
Normal ciliary beat frequency is 1000-1500 beats per minute. The frequency is slower in the peripheral airways (eg, bronchioles) compared to the larger airways (eg, trachea). The ciliary motility is maintained in the same plane along the length of airways and results in mucociliary transport rates up to 20-30 mm/min.
Pathophysiology
Defects in the ciliary component cause abnormal ciliary movements, resulting in impaired mucociliary clearance and manifesting as recurrent and or persistent sinopulmonary infections, among other problems.
Dynein arm defects manifest as a total or a partial absence of either both inner or both outer dynein arms or involve just the inner or outer arms. Sometimes, shortened dynein arms are the only defect. Recent studies show differential functions of both inner and outer dynein arms and correlate ciliary beat frequency directly with the number of outer dynein arms. The ciliary beat frequency is not correlated with the number of inner dynein arms.
Radial spoke defects exhibit either a total absence of radial spokes or an absence of radial spoke heads. These defects are easily recognized by an eccentric position of the central pair of microtubules that are normally stabilized in a central position by radial spokes. Microtubular transposition defects occur in the form of absence of the central pair of tubules with transposition of the outer doublet to the center. Other defects, such as ciliary aplasia, ciliary disorientation, [8] malaligned central pair of microtubules in adjacent cilia, and basal body abnormalities may occur after viral infections, making it unclear if they are primary or secondary defects.
Moreover, in some patients with typical clinical manifestations of PCD and low levels of nasal nitric oxide, the ciliary ultrastructure may appear normal, suggesting functional abnormalities because of other defects in ciliary components. This includes few patients with biallelic mutations in DNAH11. [9] An association has been reported between genetic mutations and ciliary ultrastructure defects, as follows: [10]
-
Outer dynein arm (ODA) defects - DNAH5, DNA11, DNA12, DNAL1, TXNDC3, CCDC114, ARMC4
-
ODA and IDA defects -LRRC50/DNAAF1, KTU/DNAAF2, DNAAF3, CCDC103, HEATR2, LRRC6, ZMYND10,DYX1C1
-
Central microtubular pair abnormalities -RSPH4A, RSPH9, RSPH1
-
Radial spoke defects -CCDC39, CCDC40
-
Normal cilia or subtle ultrastructural abnormalities -DNAH11, HYD1N, CCDC 164, CCDC65
Studies have confirmed that ciliary beat pattern is associated with specific ultrastructural defects in PCD. [11] New high-resolution digital high-speed video (DHSV) imaging has allowed the precise beat pattern of cilia to be viewed in 3 different planes in slow motion or frame-by-frame. Using this technique, 3 patterns were identified and correlated with ultrastructural defects.
In the first pattern, the cilia are virtually immotile with occasional slow, low-amplitude, stiff flickering motion. This is associated with either a combined inner and outer dynein arm defect or isolated outer dynein arm defect. In the second pattern, the cilia have stiff planar forward-backward motion with markedly reduced amplitude, a pattern associated with either an isolated inner dynein arm defect or a radial spoke defect. In the third pattern, the cilia beat in a large circular gyrating motion about the base of the cilium. This pattern is associated with transposition defect.
In patients showing moderate alteration in ciliary beating, in contrast to the patients easily diagnosed due to demonstration of immotile cilia, quantitative analysis of ciliary beating has been proposed. [12]
Etiology
Primary ciliary dyskinesia (PCD) is a genetic disorder, and it appears to follow the autosomal recessive inheritance pattern. Two genes directly implicated in autosomal recessive PCD are DNAI1 and DNAH5, which encode for components of the outer dynein arm complex. [13, 14, 15, 16] Mutations in these genes are detected in 38% of patients with PCD. Commercial testing for these mutations is available and may help with the diagnosis.
Epidemiology
United States statistics
The prevalence of PCD is approximately 1:16,000 live births. Geographic area and consanguinity may affect the prevalence. Specific types of defects are consistent within individual families and appear to be genetically determined. Based on the autosomal recessive mode of inheritance, the probability of having subsequent children with PCD is 1:4.
International statistics
The reported frequency is 1 per 26,000-40,000 live births. However, this is likely to be an underestimate because misdiagnosis is common. [17]
A genetic database analysis by Hannah et al estimated that the overall minimum global prevalence of PCD is at least 1 in 7554 persons. The expected frequency of PCD is higher in persons of African ancestry than in most other populations. [18]
Race-, sex-, and age-related demographics
No racial predilection is reported.
No sex predilection is reported.
No particular age predilection is recognized; infants are born with this genetic disorder. Cases associated with dextrocardia and with respiratory symptoms are more likely to be diagnosed in early infancy.
Prognosis
The progression of lung disease varies and is affected by the time of diagnosis, the ability of medical treatment to control symptoms, and the prevention of complications that affect the quality of life.
Some individuals have a normal or near normal lifespan. No studies have examined the impact of current symptomatic therapies on the course of disease.
Morbidity/mortality
Morbidity includes chronic, persistent, or recurrent sinusitis, rhinitis, pneumonia, and otitis media. Male infertility is common. Evidence of female infertility is inconclusive. Progression of lung disease varies and is affected by age at diagnosis, ability of medical treatment to control the symptoms, and prevention of complications. These factors affect the quality of life. Individuals with normal or near normal lifespan have been reported. No studies have examined the impact of current symptomatic therapies on the course of disease.
-
Diagram showing the cross-section of normal cilia showing its ultrastructure. Important components are labeled.
-
Ciliary ultrastructure, Left, Normal cilium from a healthy individual in which both inner and outer dynein arms can clearly identified. Right, the absence of outer and inner dynein arms in a patient with primary ciliary dyskinesia. Image courtesy of J. Carson, PhD, University of North Carolina.







