Practice Essentials
Hypopituitarism is a partial or complete insufficiency of pituitary hormone secretion that may derive from pituitary or hypothalamic disease. The onset can be at any time of life. [1] The focus of this article is childhood-onset hypopituitarism. (See the image below.)
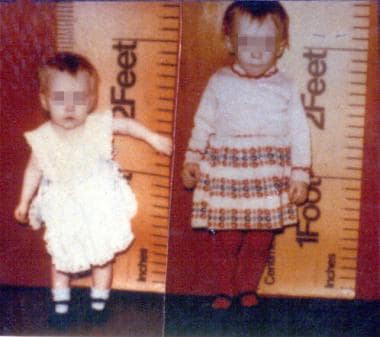 The left photograph shows an untreated 21-month-old girl with congenital hypopituitarism. The right panel depicts the same child aged 29 months, following 8 months of growth hormone therapy.
The left photograph shows an untreated 21-month-old girl with congenital hypopituitarism. The right panel depicts the same child aged 29 months, following 8 months of growth hormone therapy.
Intrinsic pituitary disease, or any process that disrupts the pituitary stalk or damages the hypothalamus, may produce pituitary hormone deficiency. The clinical presentation of hypopituitarism may vary, depending on patient age and on the specific hormone deficiencies, which may occur singly or in various combinations. As a general rule, diagnosis of a single pituitary hormone deficiency requires evaluating the other hormone axes. (See Etiology, History, and Physical Examination.)
Pituitary gland development and physiology
The pituitary gland, located at the base of the brain, is composed of anterior (ie, adenohypophysis) and posterior (ie, neurohypophysis) regions. The anterior pituitary, an ectodermal structure that derives from the pharynx as the Rathke pouch, produces most of the gland’s hormones. The major biologically active hormones released into systemic circulation from the anterior pituitary include the following:
-
Growth hormone (GH)
-
Adrenocorticotropic hormone (ACTH)
-
Thyroid-stimulating hormone (TSH)
-
Luteinizing hormone (LH)
-
Follicle-stimulating hormone (FSH)
-
Prolactin (PRL)
The pathway from embryogenesis to the full differentiation of specific functional cell types within the pituitary is controlled by numerous genes that encode transcription factors. (See the diagram below.) Mutations in these genes are causes of congenital hypopituitarism and have specific pituitary hormone deficiencies associated with the involved gene. (See Etiology.)
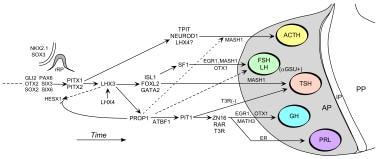 Regulation of the development of the mammalian anterior pituitary gland by transcription factors. Following, inductive signals between the developing diencephalon and the oral ectoderm, early transcription factors guide the formation of rudimentary Rathke's Pouch (rRP) and then subsequent gene regulatory pathways control the determination, proliferation, and differentiation events that establish the specialized hormone-secreting cells. AP = anterior pituitary, IP = intermediate pituitary, PP = posterior pituitary. Modified by S. Rhodes from Mullen, R.D., Colvin, S.C., Hunter, C.H., Savage, J.J., Walvoord, E.C., Bhangoo, A.P.S., Ten, T., Weigel, J., Pfäffle, R.W., and Rhodes, S.J. (2007). Roles of the LHX3 and LHX4 LIM-homeodomain factors in pituitary development. Mol. Cell. Endocrinol., 265-266: 190-195.
Regulation of the development of the mammalian anterior pituitary gland by transcription factors. Following, inductive signals between the developing diencephalon and the oral ectoderm, early transcription factors guide the formation of rudimentary Rathke's Pouch (rRP) and then subsequent gene regulatory pathways control the determination, proliferation, and differentiation events that establish the specialized hormone-secreting cells. AP = anterior pituitary, IP = intermediate pituitary, PP = posterior pituitary. Modified by S. Rhodes from Mullen, R.D., Colvin, S.C., Hunter, C.H., Savage, J.J., Walvoord, E.C., Bhangoo, A.P.S., Ten, T., Weigel, J., Pfäffle, R.W., and Rhodes, S.J. (2007). Roles of the LHX3 and LHX4 LIM-homeodomain factors in pituitary development. Mol. Cell. Endocrinol., 265-266: 190-195.
The anterior pituitary is primarily regulated by neuropeptide-releasing and release-inhibiting hormones produced in the hypothalamus. These regulatory hormones are transported to the anterior pituitary via the pituitary portal system circulation. The release-stimulating hormones produced by the hypothalamus include the following:
-
Growth hormone–releasing hormone (GHRH)
-
Corticotropin-releasing hormone (CRH)
-
Thyrotropin-releasing hormone (TRH)
-
Gonadotropin-releasing hormone (GnRH)
PRL secretion is distinct from that of the other anterior pituitary hormones, being inhibited by hypothalamic dopamine. In addition, antidiuretic hormone (ADH) produced in the hypothalamus acts synergistically with CRH to promote ACTH release.
A negative feedback loop occurs such that the hormones produced in the target glands feed back to inhibit the release of their respective regulatory pituitary and hypothalamic factors. For example, hypothalamic TRH stimulates TSH release, which in turn stimulates the thyroid gland, resulting in increased serum levels of thyroxine (T4) and triiodothyronine (T3). When they have reached sufficient levels, T3 and T4 suppress TRH and TSH release.
The posterior pituitary consists of neural tissue that descends from the floor of the third ventricle. In contrast to the anterior pituitary hormones, the posterior pituitary hormones (ie, ADH, oxytocin) are synthesized by cell bodies in the hypothalamus and transported along the neurohypophyseal tract of the pituitary stalk. Release of these hormones occurs in response to neurohypophyseal stimuli.
Etiology
Hypopituitarism has multiple possible etiologies either from congenital or acquired mechanisms. The common endpoint is disrupted synthesis or release of 1 or more pituitary hormones, resulting in clinical manifestations of hypopituitarism.
Genetic causes of hypopituitarism are relatively rare. However, research since the late 20th century has brought considerable advances in the understanding of the various genetic causes of congenital hypopituitarism. Inheritance patterns may be autosomal recessive, autosomal dominant, or X-linked recessive. The phenotype and severity of clinical findings in congenital hypopituitarism are determined by the specific genetic mutation. [2]
Mutations in pituitary transcription factors can cause multiple pituitary hormone deficiencies (MPHD) or, less commonly, an isolated pituitary hormone deficiency. Mutations in PIT1 (POUF1) [3] and PROP1 [4, 5] (ie, prophet of Pit-1) were the first mutations shown to cause MPHD. Mutations in the PIT1 gene produce a phenotype consisting of deficiencies of GH, PRL, and TSH.
PROP1 is expressed before Pit-1 and is a prerequisite for the expression of Pit-1. Inactivating mutations of PROP1 cause deficiencies of LH, FSH, GH, PRL, and TSH. Clinical phenotypes of patients with MPHD with PROP1 defects, determined by the pattern of hormone deficiency, can vary considerably even among patients with the same mutation. [6, 7]
Mutations in genes LHX3 and LHX4, which are expressed prior to Pit-1 and PROP1, have also been described as causing a phenotype of MPHD. [8] Homozygous inactivating mutations in HESX1 produce a complex phenotype with pituitary hypoplasia that resembles septo-optic dysplasia (SOD). [9] Most cases of SOD remain sporadic without a known genetic defect, and much remains to be learned about the role of HESX1 in other forms of hypopituitarism. Mutations in the GH gene or the GHRH gene lead to isolated GH deficiency (GHD). Mutations in the genes KAL and KISS1R lead to isolated gonadotropin deficiency. KAL plays a causative role in some forms of Kallmann syndrome.
Causes of hypopituitarism can be divided into categories of congenital and acquired causes. An overview of causes based on categories is summarized below.
Congenital etiologies
Congenital causes of hypopituitarism include the following:
-
Perinatal insults (eg, traumatic delivery, birth asphyxia)
-
Interrupted pituitary stalk [10]
-
Absent or ectopic neurohypophysis
-
Pallister-Hall syndrome (Hypothalamic hamartoma and polydactyly)
Genetic disorders causing hypopituitarism include the following:
-
Isolated GH deficiency types IA, IB, II, III
-
MPHD [Multiple Pituitary Hormone Deficiency] (eg, from PIT1 and PROP1 mutations)
-
Septo-optic dysplasia
-
Isolated gonadotropin deficiency (eg, from KAL and KISS1R mutations)
Developmental central nervous system (CNS) defects that cause hypopituitarism include the following:
-
Anencephaly
-
Holoprosencephaly [brain fails to divide properly into right and left hemispheres)
-
Pituitary aplasia or hypoplasia
Acquired etiologies
Cranial irradiation and hemochromatosis can lead to hypopituitarism. Delayed presentation of pituitary hormone deficiencies from radiation-induced damage [11] resulting from therapy for CNS and non-CNS tumors has received increased recognition. (See the chart below.) There is also now a recognition of immediate and delayed endocrine sequelae in survivors of traumatic brain injury. [12, 13, 14] A review [15] addresses endocrine dysfunction following pediatric traumatic brain injury.
In the era of use of biologics for cancer treatment, drug-induced hypophysitis and hypopituitarism must be considered as well. [16] Hypophysitis is a common endocrinologic adverse effect that has been attributed to immune checkpoint inhibitors. [1]
Infiltrative disorders that can cause hypopituitarism include the following:
-
Lymphocytic hypophysitis
Tumors (eg, sellar, suprasellar, pineal) that can result in hypopituitarism include the following:
-
Germinoma [18]
-
Pituitary adenoma (rare prior to adulthood)
Epidemiology
MPHD is rare in childhood, with a possible incidence of fewer than 3 cases per million people per year. The most common pituitary hormone deficiency, GHD, is much more frequent; a US study reported a prevalence of 1 case in 3480 children. [19] A 2001 population study in adults in Spain estimated the annual incidence of hypopituitarism at 4.2 cases per 100,000 population. [20] The estimated incidence of congenital hypopituitarism is 1 in 4000 to 10,000. [21]
Because hypopituitarism has congenital and acquired forms, the disease can occur in neonates, infants, children, adolescents, and adults.
Prognosis
With appropriate treatment, the overall prognosis in hypopituitarism is very good. Sequelae from episodes of severe hypoglycemia, hypernatremia, or adrenal crises are among potential complications. Long-term complications include short stature, osteoporosis, increased cardiovascular morbidity/mortality, and infertility. Previous findings of increased cardiovascular morbidity and decreased life expectancy in adults with hypopituitarism were thought to be largely secondary to untreated GHD.
Morbidity/mortality
Morbidity and mortality statistics generally cannot be viewed in isolation but must instead be related to the underlying cause of hypopituitarism. For example, morbidity and mortality are minimal in the context of idiopathic GHD compared with hypopituitarism caused by craniopharyngioma.
Recognition of pituitary insufficiency and appropriate hormone replacement (including stress doses of hydrocortisone, when indicated) are essential for the avoidance of unnecessary morbidity and mortality. Clinical manifestations of isolated or multiple deficiencies in pituitary hormones (anterior and/or posterior) can result in significant sequelae that include any of the following:
-
Hypoglycemia - Can cause convulsions; persistent, severe hypoglycemia can cause permanent CNS injury.
-
Adrenal crisis - Can occur during periods of significant stress, from ACTH or CRH deficiency; symptoms include profound hypotension, severe shock, and death.
-
Short stature - Can have significant psychosocial consequences.
-
Hypogonadism and impaired fertility - From gonadotropin deficiency
-
Osteoporosis - Results in increased fracture risk as an adult
GHD is believed to be an important contributing factor to morbidity and mortality associated with hypopituitarism. In a 2008 study, childhood onset GHD was associated with an increased hazard ratio for morbidity of greater than 3.0 for males and females. [22] Causes of morbidity and mortality are multifactorial and relate to the specific cause of hypopituitarism, as well as to the degree of pituitary hormone deficiency.
Patient Education
Teaching patients when and how to administer appropriate stress doses (oral and parenteral) of hydrocortisone is essential. When treatment includes recombinant human growth hormone (rhGH) therapy, instruct parents and patients to recognize and report adverse effects.
Express the importance of wearing a medical identification bracelet or necklace. Genetic counseling with parents and patients about the mode of transmission of hypopituitarism is important for cases involving heritable forms of the disease.
-
The left photograph shows an untreated 21-month-old girl with congenital hypopituitarism. The right panel depicts the same child aged 29 months, following 8 months of growth hormone therapy.
-
Regulation of the development of the mammalian anterior pituitary gland by transcription factors. Following, inductive signals between the developing diencephalon and the oral ectoderm, early transcription factors guide the formation of rudimentary Rathke's Pouch (rRP) and then subsequent gene regulatory pathways control the determination, proliferation, and differentiation events that establish the specialized hormone-secreting cells. AP = anterior pituitary, IP = intermediate pituitary, PP = posterior pituitary. Modified by S. Rhodes from Mullen, R.D., Colvin, S.C., Hunter, C.H., Savage, J.J., Walvoord, E.C., Bhangoo, A.P.S., Ten, T., Weigel, J., Pfäffle, R.W., and Rhodes, S.J. (2007). Roles of the LHX3 and LHX4 LIM-homeodomain factors in pituitary development. Mol. Cell. Endocrinol., 265-266: 190-195.
-
Summary of Neuroendocrine Dysfunction following radiotherapy (courtesy of Stephen M Shalet, MD)



