Background
Hyperphenylalaninemia (HPA) refers to a spectrum of disorders characterized by abnormally elevated levels of phenylalanine (PHE) in the blood (> 2mg/dl or 120 µmol/L). HPA is the commonest error of amino acid metabolism occuring in humans. [1] Traditionally, the term "hyperphenylalaninemia" has been used to refer to the milder form of the clinical condition, whereas classic phenylketonuria (PKU) is known to represent the more severe end of this spectrum. [2] Classic PKU is associated with PHE levels that exceed 20 mg/dL (1200 µmol/L) and is described separately.(see Phenylketonuria)16 A consensus statement from the NIH in 2000 classified individuals with untreated PHE levels that are above normal but below 20 mg/dl as suffering from hyperphenylalaninemia. [3]
In 2014, the American College of Medical Genetics and Genomics released a practice statement stating that the hyperphenylalaninemias represent a wide sprectrum of disease.17 HPA commonly is categorized on the basis of serum PHE levels as follows [4] :
-
Benign mild HPA: 2- 6 mg/dl
-
Mild HPA: 6-10mg/dl
-
Mild phenylketonuria (PKU): 10- 15mg/dl
-
Moderate PKU: 15-20 mg/dl
-
Classic PKU: > 20mg/dl
Phenylalanine levels of 6 mg/dL or less in patients consuming an unrestricted diet generally are considered to be a benign condition. No dietary phenylalanine restrictions usually are recommended for patients with benign mild HPA. Individuals with PHE levels >10mg/dL require treatment. Some US centers will treat infants with levels in the 6-10mg/dL range, but evidence regarding the efficacy of this approach is mixed. [5]
Pathophysiology
Phenylalanine is an essential amino acid, which means it must be obtained through the diet. In individuals with HPA, the ability to metabolize ingested phenylalanine is impaired due to defects in specific enzymes or cofactors involved in its metabolism.
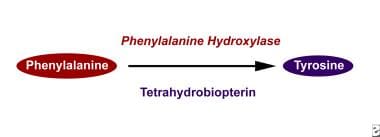 Phenylalanine hydroxylase converts phenylalanine to tyrosine.
Phenylalanine hydroxylase converts phenylalanine to tyrosine.
Most commonly, HPA is due to a deficiency of the enzyme phenylalanine hydroxylase (PAH); hence the condition is also known as phenylalanine hydroxylase deficiency. [6] The severity of the condition depends on the degree of enzyme deficiency. Classic PKU is associated with a near total absence of the enzyme. Approximately 2% of individuals with elevated phenylalanine levels have normal PAH activity but lack tetrahydrobiopterin, a crucial cofactor (BH4 Deficiency (Tetrahydrobiopterin Deficiency).
The PAH gene, located on chromosome 12q23.1 encodes the enzyme phenylalanine hydroxylase, and is the primary gene associated with hyperphenylalaninemia (HPA) and phenylketonuria (PKU). There are over 1,000 identified mutations in the PAH gene known to be associated with these conditions. These mutations can vary greatly in their nature and effects as follows:
-
Phenotype-Genotype Correlation and mutation severity Impact: The PKU phenotype's severity is largely dependent on the residual activity of the enzyme. Mutations enabling 1-5% of the enzyme's normal activity usually are associated with milder HPA cases, while those reducing enzyme activity to below 1% result in more severe PKU cases.
The clinical effects of phenylalanine hydroxylase deficiency can be summarized as follows:
-
Phenylalanine buildup: The lack of enzymatic activity leads to the accumulation of phenylalanine in the blood and tissues. Elevated phenylalanine levels are harmful, especially to the brain, and can lead to intellectual disability, behavioral issues, seizures, and other neurological complications without proper treatment.
-
Reduced tyrosine production: The enzyme phenylalanine hydroxylase normally converts phenylalanine into tyrosine. Therefore, its deficiency results in decreased tyrosine levels. Tyrosine is essential for synthesizing various vital neurotransmitters (such as dopamine, norepinephrine, and epinephrine) and melanin, which is responsible for the pigmentation in hair and skin.
-
Alternate metabolic pathways: As phenylalanine accumulates, it is metabolized through other pathways, producing phenylketones, including phenylacetate, phenylpyruvate, and phenyllactate. These substances are eliminated in the urine, and are responsible for imparting a distinct musty smell, a hallmark of PKU. In less severe cases of HPA, there's no significant buildup of phenylketones, hence they are also called non-phenylketonuric hyperphenylalaninemias.
Inheritance:
HPA is an autosomal recessive condition. Individuals develop the condition because each of their parents passed down a abnormal PAH gene to them. Only individuals with two dysfuctional PAH genes—one from the mother and one from the father—develop HPA.
-
Individuals with one functional and one non functional PAH gene are called carriers.
-
Carriers do not have or develop HPA. However, they have a 50 percent chance of passing down a nonworking copy of the gene to their children.
Epidemiology
Frequency
Frequency is approximately 15-75 cases per 1,000,000 births. About a third of newborns detected to have elevated phenylalanine levels via newborn screening turn out to have one of the milder forms of HPA; the rest have classic PKU [7] .
Mortality/Morbidity
Most individuals with hyperphenylalaninemia have normal life expectancy. Several studies have identified a linear relationship between the phenylalanine level and intelligence testing and performance.
Race
The incidence and severity of HPA shows notable variation across different ethnic groups and geographical areas around the world. [7]
Sex
Both sexes are equally affected because deficiency in phenylalanine hydroxylase activity is inherited as an autosomal-recessive trait. Pregnant women with phenylalanine levels that exceed 6 mg/dL risk having children with microcephaly, intellectual disability, and birth defects (eg, maternal hyperphenylalaninemia). [8]
Age
Hyperphenylalaninemia is commonly diagnosed at birth by newborn screening. Some cases in adult women have been detected using maternal screening programs or following birth of children with birth defects.
Prognosis
Prognosis depends on the severity of the condition and detection and treatment in a timely fashion. Mildly affected infants often require no treatment or minimal dietary restrictions. Most severely affected infants can have normal growth and development with appropriate adherence to treatment and follow up. [6]
Patient Education
Teach patients and parents about proper diet. Children should participate in their dietary planning as soon as they have that ability.
-
Phenylalanine hydroxylase converts phenylalanine to tyrosine.




