Background
Best disease, also termed vitelliform macular dystrophy, is typically an autosomal dominant disorder that classically presents in childhood with the striking appearance of a yellow or orange yolklike lesion in the macula. [1] Dr Franz Best, a German ophthalmologist, described the first pedigree in 1905. [2]
The lesion evolves through several stages over many years, with increasing potential for adverse visual outcome. A hallmark of the disease is a markedly abnormal electro-oculogram (EOG) in all stages of progression and in phenotypically normal carriers. [1, 3] The adult form varies, as described and shown in the image below.
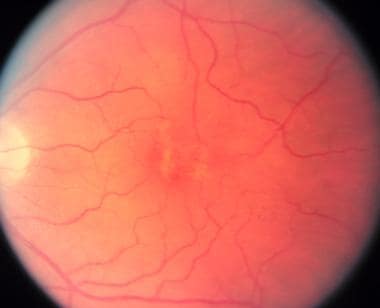 Adult vitelliform macular dystrophy resembles Best disease, but it can be differentiated by its later age of onset, smaller lesion, and normal electro-oculogram testing.
Adult vitelliform macular dystrophy resembles Best disease, but it can be differentiated by its later age of onset, smaller lesion, and normal electro-oculogram testing.
Pathophysiology
Lesions in Best disease are restricted to the eye. [1] No systemic associations exist. Abnormalities in the eye result from a disorder in the retinal pigment epithelium (RPE). Best disease is an inherited retinal dystrophy that results from a mutation in the BEST1 gene and is inherited in an autosomal dominant fashion. A dysfunction of the protein bestrophin results in abnormal fluid and ion transport by the RPE. [4] Lipofuscin (periodic acid-Schiff [PAS] positive) accumulates within the RPE cells and in the sub-RPE space, particularly in the foveal area. The RPE appears to have degenerative changes in some cases, and secondary loss of photoreceptor cells can occur over time. [5] Breakdown of RPE/Bruchs membrane can allow choroidal neovascularization to develop as a late complication.
Epidemiology
Frequency
United States
Best disease is rare.
International
Best disease is rare.
Mortality/Morbidity
Visual acuity is good in the previtelliform stage. Even with the egg-yolk appearance, visual acuity is maintained in the range of 20/20 to 20/50 (6/6 to 6/15) for many years. The breakup of the vitelliform stage, leading to the scrambled egg stage, may be accompanied by visual acuity deterioration. It is the final stages of geographic RPE atrophy with possible development of choroidal neovascular membrane that is associated with further deterioration in visual acuity. [6, 7] These changes usually occur in individuals older than 40 years. Various studies have shown that most individuals retain reading and driving vision in at least 1 eye into adulthood (88% have 20/40 or better vision). Only 4% of these individuals develop vision less than 20/200 in the better eye.
Race
Best disease is most common in individuals of European ancestry but can also be found in individuals of African and Hispanic ancestry.
Sex
No known gender predilection exists.
Age
Usual onset of Best disease is from 3-15 years, with an average age of 6 years. The condition often is not detected until much later in the disease because visual acuity may remain good for many years. The atrophic stage usually occurs after age 40 years.
Prognosis
Prognosis for Best disease is mixed. Some carriers will never phenotypically express the disorder. Some individuals will never have progression beyond the earliest stages of the disease and will maintain better than 20/40 vision in both eyes. In general, most people will maintain reading vision in at least 1 eye throughout life. In one study, 88% of patients retained 20/40 or better visual acuity, and only 4% of them had 20/200 or worse visual acuity in the better eye. The deterioration of vision usually is very slow and is not significant in most individuals until after age 40 years. [8, 9, 10]
Patient Education
Genetic inheritance: Provide an explanation of autosomal dominant inheritance to the patient and family members. In genetic counseling, discuss carrier state, variable penetrance and expressivity, and implications for offspring. Recommend familial evaluation.
Occupational counseling: Discuss the patient's prognosis and the possible implications on career direction.
Routine examination: Emphasize regular examinations because changes in fundus appearance over time may elucidate the eventual prognosis. Conduct evaluation for choroidal neovascularization.
Amsler grid: Teach use of this tool to identify central visual field changes.
Low vision aids: Assistive devices may be necessary if visual acuity deteriorates. Refer to a low vision specialist or organization.
For excellent patient education resources, visit eMedicineHealth's Eye and Vision Center. Also, see eMedicineHealth's patient education article Macular Degeneration.
-
Classic egg-yolk appearance in the second (vitelliform) stage of vitelliform macular dystrophy. The 0.5-6 mm diameter yellow or orange lesion results from an accumulation of lipofuscin beneath and within the retinal pigment epithelium. This lesion is usually noted in individuals aged 3-15 years. Visual acuity is most often preserved in the 20/20 to 20/40 range.
-
The pseudohypopyon (stage 3) lesion is found in the teenage or later years. It results from a break in the retinal pigment epithelium, allowing accumulation of the yellow substance in the subretinal space with the formation of a fluid level. This fluid can shift over 60-90 minutes with positioning.
-
The atrophic stage (stage 5) may be accompanied by the deposition of pigment or choroidal neovascularization, both of which can lead to visual deterioration.
-
The scrambled egg appearance of stage 4 results from a deterioration of the uniform cystic lesion noted in stage 2 (egg-yolk appearance). At this point, the visual acuity can begin to worsen.
-
Adult vitelliform macular dystrophy resembles Best disease, but it can be differentiated by its later age of onset, smaller lesion, and normal electro-oculogram testing.
-
The fluorescein angiogram of the latter lesion reveals a transmission defect consistent with atrophic changes in the retinal pigment epithelium. This appearance also can be found in the later stages of Best disease.
-
Spectral domain optical coherence tomography demonstrates subretinal lesion with adjacent cystoid macular edema.
-
Autosomal-recessive bestrophinopathy: Atrophic central lesion and white subretinal deposits along the vascular arcades in a 14-year-old female with 20/70 vision and no family history of Best macular dystrophy.





