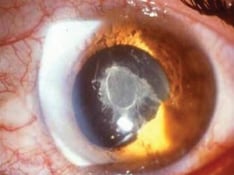
Practice Essentials
Scleral inflammation (scleritis) may occur in one or both eyes. Pain is a hallmark symptom.
Signs of scleritis include focal or diffuse redness or violaceous discoloration, initial scleral thickening, late scleral thinning, nodules, and scleral necrosis. It may be associated with keratitis, iritis, glaucoma, and exudative retinal detachment.
Scleritis commonly has an underlying cause, usually an autoimmune disease (rheumatoid arthritis, granulomatosis with polyangiitis, and other vasculitic/connective tissue diseases).
Treatment includes corticosteroids, nonsteroidal anti-inflammatory drugs (NSAIDs), immunosuppressives, and biologics.
Background
Scleritis is a chronic, painful, and potentially blinding inflammatory disease characterized by edema and cellular infiltration of the scleral and episcleral tissues (outer coat of the eye). Scleritis may be isolated to the eye but is commonly associated with systemic autoimmune disorders, including rheumatoid arthritis, systemic lupus erythematosus, relapsing polychondritis, spondyloarthropathies, Wegener granulomatosis, polyarteritis nodosa, and giant cell arteritis. [1]
Scleritis may be the initial or only clinical manifestation of these potentially lethal disorders. The correct and rapid diagnosis and the appropriate systemic therapy can halt the relentless progression of both ocular and systemic processes, thus preventing the destruction of the globe while prolonging survival and improving quality of life.
Scleritis may be classified into anterior and posterior. [2, 3] Anterior scleritis can be diffuse, nodular, necrotizing with inflammation (necrotizing), and necrotizing without inflammation (scleromalacia perforans). The most common clinical forms are diffuse scleritis and nodular scleritis. Necrotizing scleritis with or without inflammation is much less frequent, more ominous, and frequently associated with systemic autoimmune disorders and peripheral ulcerative keratitis. Posterior scleritis is characterized by flattening of the posterior aspect of the globe revealed with imaging, thickening of the posterior coats of the eye (choroid and sclera), and retrobulbar edema. [4]
Pathophysiology
An autoimmune dysregulation in a genetically predisposed host is presumed to cause scleritis. There are distinct forms of scleritis with varied pathophysiology. Inciting factors may include infectious organisms, endogenous substances, or trauma. The inflammatory process may be caused by immune complex–related vascular damage (type III hypersensitivity) and subsequent chronic granulomatous response (type IV hypersensitivity). [1]
Both immune complex vessel deposition in episcleral- and scleral-perforating capillary and postcapillary venules (inflammatory microangiopathy) and cell-mediated immune responses interact as part of the activated immune network, which can lead to scleral inflammation and destruction. The autoimmune nature of scleritis is supported by the frequent association with systemic autoimmune disorders and the favorable response to immunosuppressive therapy. [5]
Epidemiology
Frequency
United States
Although scleritis is uncommon, the exact incidence of scleritis is uncertain. The reported prevalence of scleritis is skewed by the somewhat selected referrals of the reporting institutions.
Approximately 2.6% of patients referred to the Immunology Service at the Massachusetts Eye and Ear Infirmary Hospital of Boston over 11 years had scleritis.
About 8.7% of patients referred to the Massachusetts Eye Research and Surgery Institute (MERSI) of Cambridge, MA, over 5 years had scleritis
International
Approximately 0.08% of patients referred to the Department of Ophthalmology of Southern General Hospital and Victoria Infirmary of Glasgow over 8 years had scleritis.
Mortality/Morbidity
Patients with scleritis are at risk for ocular complications and systemic disease association.
Ocular complications of scleritis, which cause vision loss and eye destruction, appear due to the extending scleral inflammation. Peripheral ulcerative keratitis (13-14%), uveitis (about 42%), glaucoma (12-13%), cataracts (6-17%), and fundus abnormalities (about 6.4%) are all common. These complications are most common in necrotizing scleritis, the most destructive type of scleritis.
Scleritis may be the first manifestation of a potentially lethal systemic disease. Disease association may be found in about 57% of patients with scleritis. Up to 48% of patients with scleritis present with a previously known connective tissue or vasculitic disease. Some diseases are potentially lethal unless treated with prompt and aggressive therapy. Other patients may present with concomitant trauma, infection, or postsurgical inflammation. Systemic disease association is most common in cases of necrotizing scleritis.
Sex
Women are more likely to have scleritis than men (1.6:1).
Age
Scleritis is most common in the fourth to sixth decades of life, with the peak incidence occurring in the fifth decade.
Prognosis
The prognosis varies depending on the disease classification. It may range from a self-limited episode of inflammation to a severe necrotizing process with vision-threatening complications.
The ocular prognosis of scleritis with systemic autoimmune diseases varies, depending on the specific autoimmune disease.
Scleritis in spondyloarthropathies or systemic lupus erythematosus, usually a relatively benign and self-limiting condition, is diffuse scleritis or nodular scleritis without ocular complications.
Scleritis in granulomatosis with polyangiitis is a severe disease that can lead to permanent blindness; it is usually necrotizing scleritis with ocular complications.
Scleritis in rheumatoid arthritis or relapsing polychondritis is a disease of intermediate severity; it may be diffuse, nodular, or necrotizing scleritis with or without ocular complications.
Scleritis without systemic disease association is often more benign than scleritis accompanied by infection or autoimmune disease. These cases of idiopathic scleritis may be mild, shorter in duration, and more likely to respond to topical steroid drops alone.
Patient Education
Patients with signs and symptoms of scleritis need to undergo an accurate eye evaluation to ensure proper treatment, determine prognosis, and exclude differential diagnoses with less vision-threatening potential.
For excellent patient education resources, visit eMedicineHealth's Eye and Vision Center. Also, see eMedicineHealth's patient education article, Eye Pain.