Overview
Modern treatment of seizures started in 1850 with the introduction of bromides, which was based on the theory that epilepsy was caused by an excessive sex drive. In 1910, phenobarbital (PHB), which then was used to induce sleep, was found to have antiseizure activity and became the drug of choice for many years. A number of medications similar to PHB were developed, including primidone.
In 1938, Houston Merrit and Tracy Putnam described animal models for screening multiple compounds for antiepileptic activity in the Journal of the American Medical Association. In 1940, phenytoin (PHT) was found to be an effective drug for the treatment of epilepsy, and since then it has become a major first-line antiepileptic drug (AED) in the treatment of partial and secondarily generalized seizures.
In 1968, carbamazepine (CBZ) was approved, initially for the treatment of trigeminal neuralgia; later, in 1974, it was approved for partial seizures. Ethosuximide has been used since 1958 as a first-choice drug for the treatment of absence seizures without generalized tonic-clonic seizures. Valproate (VPA) was licensed in Europe in 1960 and in the United States in 1978, and now is widely available throughout the world. It became the drug of choice in primary generalized epilepsies and in the mid 1990s was approved for treatment of partial seizures.
These anticonvulsants were the mainstays of seizure treatment until the 1990s, when newer AEDs with good efficacy, fewer toxic effects, better tolerability, and no need for blood level monitoring were developed. A study of live-born infants in Denmark found that exposure to the newer-generation AEDs lamotrigine, oxcarbazepine, topiramate, gabapentin, and levetiracetam in the first trimester was not associated with an increased risk in major birth defects. [1]
The new AEDs have been approved in the United States as add-on therapy only, with the exception of topiramate and oxcarbazepine (OXC); lamotrigine (LTG) is approved for conversion to monotherapy. A meta-analysis of 70 randomized clinical trials confirms the clinical impression that efficacy does not significantly differ among AEDs used for refractory partial epilepsy. [2]
Antiepileptic drugs should be used carefully, with consideration of medication interactions and potential side effects. This is particularly important for special populations, such as patients with HIV/AIDS. [3]
For more information, see Epilepsy and Seizures.
Mechanism of Action
It is important to understand the mechanisms of action and the pharmacokinetics of antiepileptic drugs (AEDs) so that these agents can be used effectively in clinical practice, especially in multidrug regimens (see the image below).

Many structures and processes are involved in the development of a seizure, including neurons, ion channels, receptors, glia, and inhibitory and excitatory synapses. The AEDs are designed to modify these processes so as to favor inhibition over excitation and thereby stop or prevent seizure activity (see the image below).
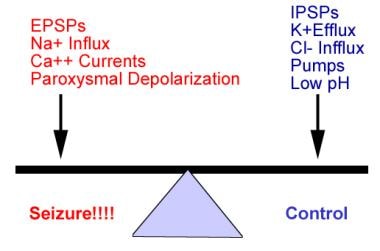 Dynamic target of seizure control in management of epilepsy is achieving balance between factors that influence excitatory postsynaptic potential (EPSP) and those that influence inhibitory postsynaptic potential (IPSP).
Dynamic target of seizure control in management of epilepsy is achieving balance between factors that influence excitatory postsynaptic potential (EPSP) and those that influence inhibitory postsynaptic potential (IPSP).
The AEDs can be grouped according to their main mechanism of action, although many of them have several actions and others have unknown mechanisms of action. The main groups include sodium channel blockers, calcium current inhibitors, gamma-aminobutyric acid (GABA) enhancers, glutamate blockers, carbonic anhydrase inhibitors, hormones, and drugs with unknown mechanisms of action (see the image below).
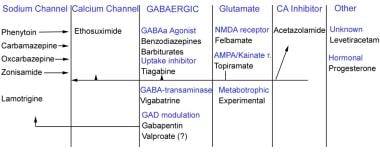 Antiepileptic drugs can be grouped according to their major mechanism of action. Some antiepileptic drugs work by acting on combination of channels or through some unknown mechanism of action.
Antiepileptic drugs can be grouped according to their major mechanism of action. Some antiepileptic drugs work by acting on combination of channels or through some unknown mechanism of action.
Sodium channel blockers
The firing of an action potential by an axon is accomplished through sodium channels. Each sodium channel dynamically exists in the following 3 states:
-
A resting state, during which the channel allows passage of sodium into the cell
-
An active state, in which the channel allows increased influx of sodium into the cell
-
An inactive state, in which the channel does not allow passage of sodium into the cell
During an action potential, these channels exist in the active state and allow influx of sodium ions. Once the activation or stimulus is terminated, a percentage of these sodium channels become inactive for a period known as the refractory period. With constant stimulus or rapid firing, many of these channels exist in the inactive state, rendering the axon incapable of propagating the action potential.
AEDs that target the sodium channels prevent the return of these channels to the active state by stabilizing them in the inactive state. In doing so, they prevent repetitive firing of the axons (see the image below).
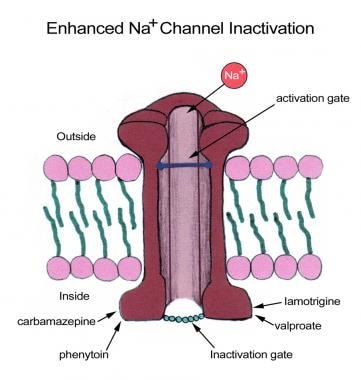 Some antiepileptic drugs stabilize inactive configuration of sodium (Na+) channel, preventing high-frequency neuronal firing.
Some antiepileptic drugs stabilize inactive configuration of sodium (Na+) channel, preventing high-frequency neuronal firing.
Calcium channel blockers
Calcium channels exist in 3 known forms in the human brain: L, N, and T. These channels are small and are inactivated quickly. The influx of calcium currents in the resting state produces a partial depolarization of the membrane, facilitating the development of an action potential after rapid depolarization of the cell.
Calcium channels function as the " pacemakers " of normal rhythmic brain activity. This is particularly true of the thalamus. T-calcium channels have been known to play a role in the 3 per second spike-and-wave discharges of absence seizures. AEDs that inhibit these T-calcium channels are particularly useful for controlling absence seizures (see the image below).
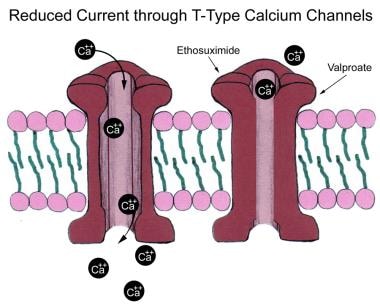 Low-voltage calcium (Ca2+) currents (T-type) are responsible for rhythmic thalamocortical spike and wave patterns of generalized absence seizures. Some antiepileptic drugs lock these channels, inhibiting underlying slow depolarizations necessary to generate spike-wave bursts.
Low-voltage calcium (Ca2+) currents (T-type) are responsible for rhythmic thalamocortical spike and wave patterns of generalized absence seizures. Some antiepileptic drugs lock these channels, inhibiting underlying slow depolarizations necessary to generate spike-wave bursts.
GABA enhancers
Gamma-aminobutyric acid (GABA) has 2 types of receptors, A and B. When GABA binds to a GABA-A receptor, the passage of chloride, a negatively charged ion, into the cell is facilitated via chloride channels (see the image below). This influx of chloride increases the negativity of the cell (ie, a more negative resting membrane potential). This causes the cell to have greater difficulty reaching the action potential. The GABA-B receptor is linked to a potassium channel.
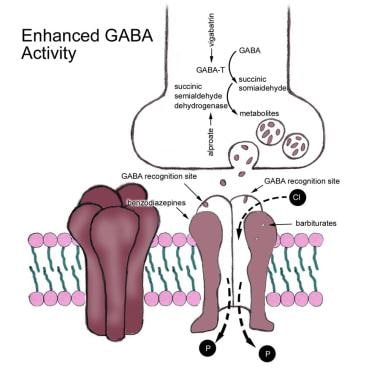 Gamma-aminobutyric acid (GABA)-A receptor mediates chloride (Cl-) influx, leading to hyperpolarization of cell and inhibition. Antiepileptic drugs may act to enhance Cl- influx or decrease GABA metabolism.
Gamma-aminobutyric acid (GABA)-A receptor mediates chloride (Cl-) influx, leading to hyperpolarization of cell and inhibition. Antiepileptic drugs may act to enhance Cl- influx or decrease GABA metabolism.
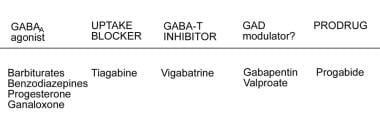
The GABA system can be enhanced by binding directly to GABA-A receptors, by blocking presynaptic GABA uptake, by inhibiting the metabolism of GABA by GABA transaminase, and by increasing the synthesis of GABA.
GABA is produced by decarboxylation of glutamate mediated by the enzyme glutamic acid decarboxylase (GAD). Some AEDs may act as modulators of this enzyme, enhancing the production of GABA and down-regulating glutamate (see the image below). Some AEDs function as an agonist to chloride conductance, either by blocking the reuptake of GABA (eg, tiagabine [TGB]) or by inhibiting its metabolism as mediated by GABA transaminase (eg, vigabatrin [VGB]), resulting in increased accumulation of GABA at the postsynaptic receptors.
Glutamate blockers
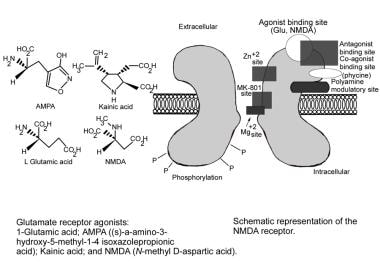
Glutamate receptors bind glutamate, an excitatory amino acid neurotransmitter. Upon binding glutamate, the receptors facilitate the flow of both sodium and calcium ions into the cell, while potassium ions flow out of the cell, resulting in excitation.
The glutamate receptor has 5 potential binding sites, as follows:
-
The alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) site
-
The kainate site
-
The N -methyl-D-aspartate (NMDA) site
-
The glycine site
-
The metabotropic site, which has 7 subunits (GluR 1-7)
AEDs that modify these receptors are antagonistic to glutamate (see the images below). Responses to glutamate antagonists differ, depending on the site being affected.
 Glutamate (main excitatory neurotransmitter in central nervous system) binds to multiple receptor sites that differ in activation and inactivation time courses, desensitization kinetics, conductance, and ion permeability. Three main glutamate receptor subtypes are N-methyl-D-aspartate (NMDA), metabotropic, and non-NMDA (alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid [AMPA] and kainate receptors). Antiepileptic drugs known to possess this mechanism of action are listed.
Glutamate (main excitatory neurotransmitter in central nervous system) binds to multiple receptor sites that differ in activation and inactivation time courses, desensitization kinetics, conductance, and ion permeability. Three main glutamate receptor subtypes are N-methyl-D-aspartate (NMDA), metabotropic, and non-NMDA (alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid [AMPA] and kainate receptors). Antiepileptic drugs known to possess this mechanism of action are listed.
Carbonic anhydrase inhibitors
Inhibition of the enzyme carbonic anhydrase increases the concentration of hydrogen ions intracellularly and decreases the pH. The potassium ions shift to the extracellular compartment to buffer the acid-base status. This event results in hyperpolarization and an increase in seizure threshold of the cells.
Acetazolamide has been used as an adjunctive therapy in refractory seizures with catamenial pattern (ie, seizure clustering around menstrual period). Topiramate and zonisamide (ZNS) also are weak inhibitors of this enzyme; however, this is not believed to be an important mechanism for their antiseizure efficacy.
Sex hormones
Progesterone is a natural anticonvulsant that acts by increasing chloride conductance at GABA-A receptors and attenuates glutamate excitatory response. It also alters messenger RNA for glutamic acid decarboxylase (GAD) and GABA-A receptor subunits. On the other hand, estrogen acts as a proconvulsant by reducing chloride conductance and acting as an agonist at NMDA receptors in the CA1 region of the hippocampus.
SV2A-binding agents
Synaptic vesicle protein 2A (SV2A) is ubiquitously expressed in the brain, but its function has not been clearly defined. SV2A appears to be important for the availability of calcium-dependent neurotransmitter vesicles ready to release their content. [4] The lack of SV2A results in decreased action potential-dependent neurotransmission, while action potential–independent neurotransmission remains normal. [5, 6]
The role of SV2A in epilepsy is confirmed by the finding that SV2A knockout mice develop a strong seizure phenotype a few weeks after birth. [5, 6] The anticonvulsant potency of SV2A ligands is correlated with their binding affinity in the audiogenic seizure-prone mice. [7, 8] Levetiracetam binds the SV2A.
Sodium Channel Blockers
Sodium channel blockade is the most common and best-characterized mechanism of currently available antiepileptic drugs (AEDs). AEDs that target sodium channels prevent the return of the channels to the active state by stabilizing the inactive form. In doing so, repetitive firing of the axons is prevented. Presynaptic and postsynaptic blockade of sodium channels of the axons causes stabilization of the neuronal membranes, blocks and prevents posttetanic potentiation, limits the development of maximal seizure activity, and reduces the spread of seizures.
Carbamazepine
Carbamazepine (CBZ) is a major first-line AED for partial seizures and generalized tonic-clonic seizures. It is a tricyclic compound and initially was used primarily for the treatment of trigeminal neuralgia; its value in the treatment of epilepsy was discovered quite by chance. CBZ’s main mode of action is to block sodium channels during rapid, repetitive, sustained neuronal firing and to prevent posttetanic potentiation. It has been approved in the United States for the treatment of epilepsy since 1974; however, it has been used for epilepsy since 1968.
CBZ is a crystalline substance that is insoluble in water and thus is limited to oral administration. It is unstable and must be protected from hot or humid conditions, which decrease its bioavailability by 50%. Approximately 75-85% of the drug is plasma protein bound, and it has a free fraction of 20-24% of the total plasma concentration. Cerebrospinal fluid (CSF) levels range from 17% to 31%. It is metabolized extensively in the liver and induces its own metabolism. The major metabolic pathway is epoxidation to CBZ 10,11-epoxide and hydrolysis to CBZ 10,11-trans -dihydrodiol.
Because CBZ induces its own metabolism, causing an increase in clearance and a decrease in levels, the serum half-life decreases by 50% during the first few weeks of treatment. The elimination half-life ranges from 5 to 26 hours following repeated treatment in healthy volunteers and patients with epilepsy. In children, the half-life ranges from 3 to 32 hours. Its induction of hepatic cytochrome P-450 system activity also increases the metabolism of other AEDs. Peak levels of the drug are present in the blood for 4-8 hours.
Formulations that are available include suspension, syrup, tablets (100 mg, 200 mg, 400 mg), chewable tablets (100 mg, 200 mg), extended-release capsules (Tegretol XR; 100 mg, 200 mg, 400 mg), Carbatrol (200 mg, 300 mg), and rectal suppositories. The extended-release preparations, Tegretol XR (Novartis) and Carbatrol (Shire), are better tolerated than the immediate-release preparations.
CBZ is one of the most widely used AEDs in the world. It is highly effective for partial-onset seizures, including cryptogenic and symptomatic partial seizures. It also has demonstrated good efficacy in the treatment of generalized tonic-clonic seizures. The drug is highly effective and well tolerated. Its major disadvantages are transient adverse dose-related effects at initiation of therapy and occasional toxicity.
Potential dose-related adverse effects include dizziness, diplopia, nausea, ataxia, and blurred vision. Rare idiosyncratic adverse effects include aplastic anemia, agranulocytosis, thrombocytopenia, and Stevens-Johnson syndrome. Asymptomatic elevation of liver enzymes is observed commonly during the course of therapy in 5-10% of patients. Rarely, severe hepatotoxic effects can occur.
Several drugs, such as macrolide antibiotics (eg, erythromycin and clarithromycin), isoniazid, chloramphenicol, calcium channel blockers, cimetidine, and propoxyphene (withdrawn from the US market), inhibit the hepatic enzyme cytochrome P-4503A4 (CYP3A4), which is responsible for the metabolic breakdown of CBZ, thereby raising its levels.
Phenobarbital (PHB), phenytoin (PHT), felbamate, and primidone also lower CBZ levels through CYP3A4. Toxic symptoms or breakthrough seizures may occur if the dose of CBZ is not adjusted. Grapefruit juice and St. John’s wort are inducers of CYP3A4 and can decrease CBZ levels.
CBZ induces the metabolism of tricyclic antidepressants, oral contraceptives, cyclosporin A, and warfarin. Any drug that is metabolized by the hepatic enzyme CYP3A4 will have reduced levels because CBZ induces this enzyme.
Phenytoin
Since 1938, phenytoin (PHT) has been a major first-line AED in the treatment of partial and secondary generalized seizures in the United States. It blocks movements of ions through sodium channels during propagation of the action potential and thus blocks and prevents posttetanic potentiation, limits development of maximal seizure activity, and reduces the spread of seizures. It also has an inhibiting effect on calcium channels and the sequestration of calcium ions in nerve terminals, thereby inhibiting voltage-dependent neurotransmission at the level of the synapse.
In addition, PHT has an antiepileptic effect on calmodulin and other secondary messenger systems, the mechanisms of which are unclear. The adverse-effect profile (eg, gingival hyperplasia and coarsening of the facial features in women) makes its use less desirable than CBZ in some patients.
PHT is a lipid-soluble crystalline powder that is a weak acid and has a pKa in the range of 8.3-9.2, which makes it soluble in alkaline solutions. Usually, it is administered to patients as a sodium salt. It is not absorbed in the stomach because of the low pH of the gastric juices but is absorbed rather slowly in the small intestines, the juices of which have a higher pH. Food and diseases of the small intestines alter PHT absorption.
Oral bioavailability is approximately 95%, and peak level after oral administration is reached in approximately 4-12 hours. It is 70-95% bound to plasma protein, and the volume of distribution is 0.5-0.8 L/kg. The brain-plasma ratio is between 1 and 2. PHT is metabolized in the liver by the hepatic P-450 mixed oxidase system and follows zero-order kinetics. A number of minor metabolites are formed, but none of them are active (ie, they have no antiepileptic properties). Excretion is through the kidneys. Elimination half-life is 7-42 hours.
The drug is available as capsules (25 mg, 50 mg, 100 mg, 200 mg), chewable tablets (50 mg), suspension (30 mg/5 mL, 125 mg/5 mL), and injection (250 mg/5 mL). Administration frequency is 1-2 times a day.
PHT is one of the most commonly used first-line or adjunctive treatments for partial and generalized seizures, Lennox-Gastaut syndrome, status epilepticus, and childhood epileptic syndromes. It is not indicated for myoclonus and absence seizures. This drug is highly effective and economical for the patient; however, tolerability of the drug is still in dispute.
One disadvantage of PHT is that it causes central nervous system (CNS) and systemic adverse effects. Long-term use of PHT has been associated with osteoporosis; therefore, this agent must be used with caution in susceptible populations, and routine screening must be performed to detect the condition early. CNS effects occur particularly in the cerebellum and the vestibular system, causing ataxia and nystagmus. PHT is not a generalized CNS depressant; however, some degree of drowsiness and lethargy is present, without progressing to hypnosis.
Nausea and vomiting, rash, blood dyscrasias, headaches, vitamin K and folate deficiencies, loss of libido, hormonal dysfunction, and bone marrow hypoplasia are among the most common adverse effects. When given during pregnancy, PHT, like other AEDs, can cause cleft palate, cleft lip, congenital heart disease, slowed growth rate, and mental deficiency in the offspring.
Among all AEDs, PHT has one of the most problematic drug interaction profiles. The 2 major reasons are its highly protein-bound (>90%) nature and its use of the P-450 enzymes for metabolism. CBZ and PHB have variable and unpredictable effects (ie, increase or decrease) on PHT levels, in that they both induce and compete for hepatic enzymes. Valproate (VPA) raises levels of PHT by displacing PHT from its protein-binding site and inhibiting its metabolism.
Other drugs that significantly increase PHT levels are isoniazid, cimetidine, chloramphenicol, dicumarol, and sulfonamides. Drugs that lower PHT levels are vigabatrin (VGB) and amiodarone.
PHT itself is a strong inducer of hepatic enzymes and alters levels of other drugs. It decreases levels of CBZ, ethosuximide, felbamate, primidone, tiagabine (TGB), and PHB. It inhibits dicumarol, warfarin, and corticosteroids; clotting factors and immunosuppression must be monitored and doses adjusted accordingly. Other drugs whose levels are reduced by PHT and require monitoring and adjustment include furosemide, cyclosporin, folate, and praziquantel. levels of chloramphenicol and quinidine are elevated by PHT.
Because of PHT’s poor adverse-effect profile, epileptologists generally try to avoid prescribing it. Despite the difficult pharmacokinetics and the adverse effects, this drug is still used widely. The once-daily dosing, the good efficacy, the extensive experience amassed, the possibility of monitoring the plasma levels, and the availability of a parenteral preparation make PHT suitable for use by the primary care physician.
Fosphenytoin
Fosphenytoin sodium is a prodrug intended for parenteral administration. Its active metabolite is PHT. It is safer and better tolerated than PHT and can be infused 3 times faster than intravenous (IV) PHT can.
When administered by IV infusion, maximum plasma fosphenytoin concentration is achieved at the end of the infusion. Fosphenytoin is completely bioavailable after intramuscular (IM) administration. Peak concentration occurs at 30 minutes after administration. The half-life is 15 minutes, and the drug is presumed to be metabolized completely by phosphatases to PHT. Fosphenytoin is not excreted in the urine.
The antiepileptic effect of fosphenytoin is attributable to its active metabolite, PHT. It is clearly better tolerated than PHT. One double-blind controlled study compared the infusion tolerance of fosphenytoin at 150 mg/min and PHT 50 mg/min. Local intolerance was reported in 9% of patients after fosphenytoin loading; 21% had infusion disrupted, and the infusion time was 13 minutes. In patients who received PHT, on the other hand, local intolerance was reported in 90%, 67% had infusion disrupted, and infusion time averaged 44 minutes.
Fosphenytoin is indicated for treatment of status epilepticus and for short-term parenteral administration when other routes are not available or inappropriate.
Cardiovascular depression and hypotension may occur with fosphenytoin but to a lesser extent than with PHT. These adverse effects usually are related to the rate of infusion. Slower infusion is recommended in susceptible patients. Severe burning, itching, and/or paresthesia, mainly in the groin area, have been associated with rapid infusion. The discomfort may be improved by lowering the infusion rate or temporary discontinuation. Hepatic or hemopoietic adverse reactions, like those seen with PHT, also may occur.
Fosphenytoin is a better IV preparation than PHT, mainly because of tolerability and safety. It also may allow faster achievement of therapeutic serum PHT levels. However, fosphenytoin is much more expensive than PHT.
Oxcarbazepine
Oxcarbazepine (OXC) is a recently developed analogue of CBZ. It was developed in an attempt to maintain the benefits of CBZ while avoiding its auto-induction and drug interaction properties. Licensed in over 50 countries, including the United States, OXC now is considered a first-line therapy in some countries. [9, 10, 11, 12]
OXC does not produce the epoxide metabolite, which is largely responsible for the adverse effects reported with CBZ. Like CBZ, OXC blocks the neuronal sodium channel during sustained rapid repetitive firing.
OXC is absorbed almost completely on oral administration and can be taken with food. It is metabolized to the active 10-monohydroxy metabolite (MHD), 10,11-dihydro-10-hydroxy-5H-dibenz[b,f]azepine-5-carboxamide. MHD is the active compound that is responsible for the pharmacologic effects of OXC.
Volume of distribution is 0.3-0.8 L/kg. Protein binding is 38%. The drug readily crosses the blood-brain barrier. Metabolism takes place in the liver; no epoxide is formed, accounting for the better tolerability of this drug than of CBZ. It induces some cytochrome P-450 enzymes, including CYP3A4, CYP3A5, and CYP2C19, but other cytochrome enzymes appear to be unaffected. Excretion is via the kidneys; peak levels are reached in 4 hours. The half-life is 8-10 hours.
OXC interacts with oral contraceptives, thereby reducing their efficacy. It does not increase the metabolism of warfarin, cimetidine, erythromycin, verapamil, or dextropropoxyphene.
OXC is approved for monotherapy or adjunctive therapy in patients with partial and secondary generalized seizures. It is an effective drug for partial seizures but may aggravate myoclonic or absence seizures. Four randomized, double-blind trials of this agent as monotherapy demonstrated effectiveness superior to that of placebo in patients with refractory epilepsy and in candidates for epilepsy surgery.
OXC is better tolerated and has fewer drug interactions than CBZ. Retrospective studies have reported worsening of seizures caused by oxcarbazepine in juvenile idiopathic generalized epilepsies. Substitution for CBZ can be made abruptly with an OXC-to-CBZ ratio of 300:200. Comparison studies of tolerability between slow-release CBZ and OXC are not available.
Available formulations are tablets (150 mg, 300 mg, 600 mg), and the recommended frequency of administration is twice a day. The initial dose in children is 10 mg/kg/d, with titration up to a maximum of 30 mg/kg. In adults, the dose is 600 mg/d up to a maximum of 2400 mg/d. Some patients require low starting doses (300 mg/d) and slower titration for better tolerability.
Somnolence, headache, dizziness, rash, hyponatremia, weight gain, gastrointestinal (GI) disturbances, and alopecia are the most commonly reported adverse effects. The allergic rash is similar to the one caused by CBZ. Dose-related adverse effects include fatigue, headache, dizziness, and ataxia. Hyponatremia is mild and can be corrected by fluid restriction. Hyponatremia is uncommon in children younger than 17 years, but it occurs in 2.5% of adults and 7.4% of the elderly. Idiosyncratic reactions appear to be less common than with CBZ.
Eslicarbazepine
Eslicarbazepine acetate (Aptiom) is a prodrug that is activated to eslicarbazepine (S-licarbazepine), the major active metabolite of oxcarbazepine. It is indicated as either adjunctive treatment or monotherapy for partial-onset seizures in adults and children 4 years and older.
The initial adult dose is 400 mg PO once daily for 1 week, then increased to 800 mg PO once daily (the recommended maintenance dose). Some patients may benefit from 1,200 mg/day (maximum dose). An increase to 1,200 mg/day should only be initiated after patients tolerate 800 mg/day for 1 week. For some patients, treatment may be initiated at 800 mg/day if the need for additional seizure reduction outweighs an increased risk of adverse reactions during initiation. It may be administered with or without food.
An increased dose may be needed if coadministered with CYP enzyme-inducing AEDs (eg, carbamazepine, phenytoin, phenobarbital, primidone). Dosage reduction is recommended with moderate-to-severe renal impairment (200 mg/day initially for 2 weeks, then increase to 400 mg/day).
In September 2017, FDA expanded indication for eslicarbazepine acetate (Aptiom) to include the treatment of partial-onset seizures (POS) in children and adolescents aged 4–17 years. The expanded approval for eslicarbazepine acetate was based on the FDA guidance that allows data extrapolation to support pediatric use. The efficacy and safety of monotherapy and adjunct therapy for adults with POS was previously established in 5 multicenter, randomized, controlled studies. This data also supported safety and tolerability of eslicarbazepine acetate for the treatment of pediatric patients with POS.
Common adverse effects (ie, >10%) include dizziness, somnolence, nausea, headache, and diplopia.
Lamotrigine
Lamotrigine (LTG) is a triazine compound that is chemically unrelated to any of the other AEDs. It was developed as an antifolate agent on the basis of a theory that the mechanism of some AEDs is related to their antifolate property. LTG was approved in the United States in 1994. [13, 14, 15]
LTG’s major mechanism of action is blocking voltage-dependent sodium-channel conductance. It has been found to inhibit depolarization of the glutaminergic presynaptic membrane, thus inhibiting release of glutamate. It has a weak antifolate effect that is unrelated to its antiseizure efficacy.
On oral administration, LTG has a bioavailability close to 100%, reaching peak levels within 1-3 hours and achieving a volume of distribution of 0.9-1.3 L/kg. Its solubility is poor in both ethanol and water; therefore, it is not available in parenteral form. Protein binding is 55% and the elimination half-life is 24-41 hours. It is metabolized by the liver and excreted through the kidneys. It produces auto-induction at higher doses and has no active metabolites.
LTG levels increase with concomitant use of valproate (VPA) to 70 hours. Combination therapy with VPA enhances the antiepileptic effect; however, it also increases the chances of developing allergic skin reactions. LTG does not induce or inhibit hepatic enzymes; therefore, it does not affect the metabolism of lipid-soluble drugs such as warfarin and oral contraceptives. Conversely, drugs that induce hepatic enzymes may reduce the half-life of LTG from 23 hours to 14-16 hours. LTG levels must be adjusted accordingly.
LTG’s significant effect on seizures was demonstrated in 9 of 10 placebo-controlled trials in which LTG was administered as add-on therapy. LTG resulted in a 17-59% reduction in seizures, with most trials showing 25-30% median reduction in seizures.
LTG is effective in partial onset and secondarily generalized tonic-clonic seizures, primary generalized seizures (ie, absence seizures and primary generalized tonic-clonic seizures), atypical absence seizures, tonic/atonic seizures, and Lennox-Gastaut syndrome. It is sometimes effective for myoclonic seizures but can cause worsening of myoclonic seizures in some patients with juvenile myoclonic epilepsy or myoclonic epilepsy of infancy.
LTG currently is approved in the United States for adjunctive therapy for partial onset and secondarily generalized tonic-clonic seizures, crossover to monotherapy, and Lennox-Gastaut syndrome.
The dose regimen and titration schedule depends on coadministration of other AEDs, the titration rate being slower with enzyme-inhibiting AEDs such as valproate than with enzyme-inhibiting AEDs such as PHT and CBZ.
Preset packages are available with the recommended doses of LTG, with and without VPA. In children on VPA, the starting dose of LTG is 0.15 mg/kg, with increments every 1-2 weeks up to a maximum of 1-5 mg/kg. In patients taking concomitant enzyme inducers, the starting dose is 0.6 mg/kg, up to a maximum of 5-15 mg/kg. LTG is available in tablets (25 mg, 50 mg, 100 mg, 150 mg, and 200 mg) and chewable tablets (5 mg, 25 mg, and 100 mg); it is administered twice a day.
Unlike most AEDs, LTG produces few CNS side effects. Rash is the main concern associated with this drug; it occurs in 5% of patients and is associated with rapid titration. Severe rash (more common in children taking VPA) may develop and lead to Stevens-Johnson syndrome, which may be fatal (though this is rare, with an incidence of only 0.1%). Other commonly reported adverse reactions are headache, blood dyscrasias, ataxia, diplopia, GI disturbance, psychosis, tremor, hypersensitivity reactions, somnolence, and insomnia.
LTG is the only AED with more than 500 documented pregnancy exposures. The International Lamotrigine Pregnancy Registry Update reported 414 monotherapy exposures, giving a risk of 2.9%. The North American AED Pregnancy Registry found no overall risk of major malformations in 684 infants exposed to LTG monotherapy but noted an increased risk of orofacial clefts. In contrast, the EUROCAT congenital anomaly registers did not find an increased risk of orofacial clefts for 40 children exposed to LTG monotherapy. [16]
The excellent side-effect profile and lack of significant CNS toxicity make this drug one of the preferred choices in treating elderly patients. The reported low incidence of congenital malformations when exposed to pregnant patients makes this drug one of the preferred treatments during pregnancy.
Zonisamide
Zonisamide (ZNS) was synthesized as a benzisoxazole in 1974. It is chemically unrelated to any of the other AEDs; it is a small-ringed structure related to sulfonamide antibiotics with pH-dependent solubility in water. [17, 18, 19, 20, 21, 22, 23, 24]
The major mechanism of action of ZNS is reduction of neuronal repetitive firing by blocking sodium channels and preventing neurotransmitter release. It also exerts influence on T-type calcium channels and prevents influx of calcium. In addition, ZNS exhibits neuroprotective effects through free radical scavenging.
When administered orally, ZNS is absorbed quickly and completely, reaching peak levels in 2-4 hours. It has a relatively long half-life of 60 hours. It has a high affinity for binding to red blood cells (RBCs) and a 40% protein-binding capacity, exhibiting a linear dose/plasma concentration at doses of 100-400 mg.
Partially metabolized by the liver (70%), ZNS uses the cytochrome P-450 system, which is followed by glucuronidation. Although it uses the cytochrome P-450 system, it is not an inducer of the system. Metabolites of ZNS are not biologically active, and 35% of the drug is excreted unchanged in the urine.
ZNS has been approved by the US Food and Drug Administration (FDA) as adjunctive therapy for patients with partial seizures who are 12 years or older. It is preferred clinically because of the ease of patient tolerance, degree of seizure reduction, long half-life, and lack of drug interactions with other AEDs. ZNS provides dose-dependent, effective, and generally well-tolerated adjunctive therapy in patients with partial seizures.
Retrospective studies have shown that ZNS is a very effective treatment for myoclonus, especially in juvenile myoclonic epilepsy. In small series of women of childbearing years, spontaneous abortions and congenital abnormalities in human fetuses have been reported at a rate of 7%, which is more than twice the rate in the general population (2-3%). However, many of these women were treated with polytherapy.
The most commonly reported adverse reactions to ZNS are dizziness, anorexia, headache, ataxia, confusion, speech abnormalities, mental slowing, irritability, tremor, and weight gain. Gradual titration of the drug appears to reduce the manifestations of adverse reactions. Somnolence and fatigue have been reported frequently. ZNS is associated with renal stones in 1.5% of patients; therefore, the risk in patients with a history of renal stones must be weighed against the therapeutic benefits of the medication.
Oligohidrosis has been reported in children, mainly as a result of the effect on carbonic anhydrase. Idiosyncratic skin reactions (eg, Stevens-Johnson syndrome, toxic epidermal necrolysis) have been reported in Japan at a rate of 46 per million patient-years of exposure. ZNS should not be used in patients who are allergic to sulfonamides.
PHT, CBZ, PHB, and VPA decrease the half-life from 63 hours to 27-46 hours, thereby reducing levels of ZNS; however, ZNS does not affect the levels of these drugs.
ZNS is a good alternative for patients with compliance problems because of its long half-life; it can be used once daily without significant fluctuation of blood levels. In addition, it does not have the cosmetic and pharmacokinetic problems of PHT. Its mechanism of action, inhibiting thalamic T-calcium currents, may make it effective in absence epilepsy and juvenile myoclonic epilepsy.
Lacosamide
Lacosamide (formerly known as erlosamide, harkoseride, or SPM 927) is an amino acid derivative referred to as functionalized amino acid. The (R)-enantiomer lacosamide has about twice the potency of the racemic mixture. Lacosamide is inactive against clonic seizures induced by bicuculline and picrotoxin but showed efficacy against hippocampal kindled seizures at least as much as other AEDs including phenytoin, carbamazepine, and valproate. [25] Lacosamide was approved in the United States in 2008.
Lacosamide has a novel mechanism of action of modulation of voltage-gated sodium channels by selective enhancement of slow inactivation but without apparent interaction with fast inactivation gating. [26] This effect may be relatively selective for neurons involved in a seizure activity in which the persistence of sodium currents is more pronounced and preserve the function of a relative less active neurons. [27] Lacosamide does not affect AMPA, kainate, NMDA, GABAA, GABAB, or various dopaminergic, serotoninergic, adrenergic, muscarinic, or cannabinoid receptors and does not block potassium or calcium currents. [28]
Upon oral administration, lacosamide has a bioavailability close to 100% and is not affected by food; intravenous infusion of lacosamide has demonstrated bioequivalence with the same dose of oral administration. Peak plasma levels occur approximately 1-4 hours after the dose, and elimination half-life is approximately 13 hours. Lacosamide has minimal protein binding (< 15%) and does not act as an inducer or inhibitor of the cytochrome P-450 (CYP-450) isoenzymes and does not have a significant interaction with other AEDs. [29]
Lacosamide effect on seizures was demonstrated as adjunctive therapy in partial-onset seizures in three 12-week, randomized, double-blind, placebo-controlled, multicenter trials in adult patients. [30, 31, 32] Lacosamide has been used in children with refractory epilepsy of different etiologies [33, 34] and in adults juvenile myoclonic epilepsy [35] with apparently good tolerability and fair efficacy, but controlled studies confirming the findings are lacking. Lacosamide has been used as well in patients in a critical care setting with nonconvulsive status epilepticus given the favorable pharmacokinetics and intravenous route access; [36, 37] however, the safety and efficacy of lacosamide in this situations has not been determined yet.
Lacosamide currently is approved in the United States as monotherapy and as adjunctive therapy for adults and adolescents aged 17 years or older with partial-onset seizures.
The dose regimen and titration is based on clinical response and tolerability. The starting dose for adjunctive therapy is 50 mg BID, and then increased by weekly increments of 50 mg BID up to 100-200 mg BID.
The starting dose for monotherapy is 100 mg BID initially, then increased at weekly intervals by 50 mg BID up to 150-200 mg BID. Alternatively, a 200 mg loading dose may be given, followed 12 hours later by 100 mg BID for 1 week, and then gradually increased at weekly intervals by 50 mg BID up to 150-200 mg BID.
Commonly reported adverse reactions include dizziness, headache, nausea, and diplopia.
Lacosamide has been labeled pregnancy category C because it has produced developmental toxicity (increased embryofetal and perinatal mortality, growth deficit) in rats following administration during pregnancy. No human data is currently available; however, a pregnancy registry is ongoing.
The excellent pharmacokinetic profile and relative good tolerability make this drug easy to use as add-on therapy (it is not approved as initial monotherapy in the United States). Intravenous formulation makes this drug particularly useful in ICU settings. The efficacy of lacosamide in status epilepticus has not been determined.
Cenobamate
Cenobamate was approved by the FDA in November 2019 for adults with partial-onset seizures as either monotherapy or adjunctive therapy. The precise mechanism if unknown, but it has shown to reduce repetitive neuronal firing by inhibiting voltage gated sodium currents. It is also a positive allosteric modulator of GABA-A ion channel.
Cenobamate was evaluated in patients with uncontrolled focal (partial)-onset epilepsy. The clinical trial program included over 1900 patients. Adjunctive treatment with cenobamate was found to significantly decrease seizure frequency in 2 well-controlled studies, however cases of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome were reported in early clinical development among the first patients exposed to the drug. In December 2018, results from a large Phase 3 safety study showed that among the 1037 patients exposed to cenobamate, no cases of DRESS were identified; reducing the starting dose and slowing titration rate appeared to reduce the risk.
Result from a multicenter, double-blind, randomized, placebo-controlled study (Study 013) at 107 epilepsy and neurology centers in 16 countries was conducted. The dose-response study included a 6-week titration phase followed by a 6-week maintenance phase. A statistically significant 56% reduction in median seizure frequency was seen with cenobamate 200 mg/day (n=113) compared with a 22% reduction with placebo (n=108). In Study 017, which included a 6-week titration phase followed by a 12-week maintenance phase, patients randomized to Xcopri 100 mg/day (n=108), 200 mg/day (n=109) or 400 mg/day (n=111) had statistically significant 36%, 55% and 55% reductions in median seizure frequency, respectively, compared with a 24% reduction with placebo (n=106). [38]
During the maintenance phase, a post-hoc analysis showed that 28% of patients receiving Xcopri had zero seizures, compared with 9% of placebo patients. During the maintenance phase of another study (Study 017), 4% of patients in the cenobamate 100 mg/day group, 11% of patients in the cenobamate 200 mg/day group, 21% of patients in the cenobamate 400 mg/day group and 1% of patients in the placebo group reported zero seizures. [39]
GABA Receptor Agonists
A seizure reflects an imbalance between excitatory and inhibitory activity in the brain, with an increment of excitation over inhibition. The most important inhibitory neurotransmitter in the brain is gamma-aminobutyric acid (GABA).
GABA-A receptors have multiple binding sites for benzodiazepines, barbiturates, and other substances (eg, picrotoxins, bicuculline, and neurosteroids). These drugs bind to different receptor sites to exert their action, but the clinical implications of each receptor site are not well understood.
The benzodiazepines most commonly used for treatment of epilepsy are lorazepam, diazepam, clonazepam, and clobazam. The first 2 drugs are used mainly for emergency treatment of seizures because of their quick onset of action, availability in intravenous (IV) forms, and strong anticonvulsant effects. Their use for long-term treatment is limited because of the development of tolerance.
The 2 barbiturates mostly commonly used in the treatment of epilepsy are phenobarbital (PHB) and primidone. They bind to a barbiturate-binding site of the benzodiazepine receptor to affect the duration of chloride channel opening. They have been used widely throughout the world. They are very potent anticonvulsants, but they have significant adverse effects that limit their use. With the development of new drugs, the barbiturates now are used as second-line drugs for the treatment of chronic seizures.
Clobazam
Clobazam has a 1,5 substitution instead of the usual 1,4-diazepine. This change results in an 80% reduction in its anxiolytic activity and a 10-fold decrease in its sedative effects. It has been licensed in Europe since 1975 but is not available in the United States. In addition to its agonist action at the GABA-A receptor, clobazam may affect voltage-sensitive conductance of calcium ions and the function of sodium channels.
Clobazam is relatively insoluble in water; therefore, no IV or intramuscular (IM) preparations are available. Its oral bioavailability is about 90%. Time to peak plasma concentration (Tmax) is 1-4 hours. The absorption rate is decreased when clobazam is taken with meals, but total absorption is not affected.
Plasma protein binding of clobazam is approximately 83% with the proportion of bound to unbound drug independent of clobazam concentration. Very low plasma protein levels are associated with increases in the unbound (ie, free) fraction, for example, in renal or hepatic disease. Brain and saliva concentrations are proportional to the unbound fraction. A good correlation exists between dosage and plasma levels; significant interindividual variations exist.
Clobazam is metabolized by oxidation in the liver to norclobazam (N -desmethylclobazam). This metabolite has a very long half-life (ie, 50 h), but it has a low affinity for the benzodiazepine receptor, and its antiepileptic effect is unclear. The elimination half-life usually is in the range of 10-50 hours. Norclobazam is conjugated in the liver and excreted in the bile as glucuronate and in the urine as sulfate. The clobazam plasma level is 20-350 ng/mL Norclobazam levels typically are 10 times higher than clobazam levels at usual clinical dosage.
No significant clinical interactions are reported for clobazam. Minor interactions are common.
Clobazam is a potent anticonvulsant for partial epilepsy. No double-blind, controlled studies have been reported, but the trials performed showed a striking benefit. In 1 study, the mean reduction of seizures was 50% in more than 50% of patients. These patients had partial epilepsy and were taking other antiepileptic drugs (AEDs). In 1 Canadian study in drug-naïve children, clobazam monotherapy was found to be as effective as CBZ or PHT.
The major clinical problem with clobazam is the development of tolerance; sedation tolerance is more evident than antiepileptic tolerance. No clear correlation between plasma levels and seizure control has been found. No measures have been effective against the development of tolerance. The anxiolytic effect (mild) may be beneficial for some patients. Clobazam is effective in a wide range of epilepsies and should be considered as adjunctive therapy. It can be used in patients with Lennox-Gastaut syndrome or primary or secondarily generalized seizures.
Clobazam is administered orally at a dose 10-20 mg/d, taken at night or twice daily. No parenteral preparations are available.
Essentially, the adverse effects of clobazam are similar to those of other benzodiazepines. The most common effect is sedation. Other adverse effects include dizziness, ataxia, blurred vision, diplopia, irritability, depression, muscle fatigue, and weakness. Idiosyncratic reactions are very rare and no fatal reactions have been reported so far.
Clobazam is useful in intermittent treatments (eg, catamenial epilepsy) and as prophylaxis for some situations, such as traveling, celebrations, and other occasions.
Clonazepam
Clonazepam, a 1,4-substituted benzodiazepine, was one of the first benzodiazepines to be used for epilepsy. It is employed in the treatment of all types of myoclonus and is useful in patients with concomitant anxiety disorder.
Clonazepam has higher affinity for the GABA-A receptor site than diazepam and binds to GABA-A receptors that do not bind with other benzodiazepines. It may have some action on sodium-channel conductance.
Clonazepam has an oral bioavailability of 80%. Tmax is 1-4 hours, but it could be delayed for as long as 8 hours. Plasma protein binding is 86% with a volume of distribution of 1.5- 4.4 L/kg. It is highly lipid soluble and can cross the blood-brain barrier rapidly. Plasma levels and antiepileptic effects are not correlated.
Clonazepam is acetylated in the liver; therefore, the metabolic rate depends on the genetic acetylator function. The metabolites of clonazepam have no clinical relevance. The drug has an elimination half-life ranging from 20 to 80 hours, and it has a very low clearance (approximately 100 mL/min in adults). Less than 0.5% is excreted in the urine.
Clonazepam levels are decreased by coadministration of enzyme-inducing drugs. No significant clinical interactions have been reported.
Clonazepam is a potent AED and the drug of choice for myoclonic seizures and subcortical myoclonus. It is also effective in generalized convulsions and, to a lesser extent, in partial epilepsies. It rarely is used as adjunctive treatment of refractory epilepsy because of its sedative effect and tolerance, which are similar to those of other benzodiazepines. It is very effective in the emergency treatment of status epilepticus, like diazepam, and can be given IV or rectally.
Withdrawal from clonazepam may induce status epilepticus or exacerbation of seizures. Psychiatric withdrawal also may occur, manifested as insomnia, anxiety, psychosis, and tremor.
Clonazepam is available as 0.5 mg, 1 mg, and 2 mg tablets and as an IV solution. The usual starting dosage is 0.25-4 mg/d once or twice daily. Slow titration is recommended.
Clonazepam’s major adverse effect is sedation, even at low doses. Children tolerate this medication much better than adults do; therefore, pediatricians use it most often. Clonazepam has the typical adverse effects of benzodiazepines (eg, ataxia, hyperactivity, restlessness, irritability, depression, cardiovascular or respiratory depression). Children and infants may have hypersalivation. Occasionally, tonic seizures may be exacerbated. Idiosyncratic reactions are rare and include marked leukopenia.
Midazolam intranasal
Midazolam intranasal is a very short-acting, water-soluble imidazobenzodiazepine. Solubility of midazolam is accomplished by the presence of an imidazole ring fused at positions 1 and 2 of the benzodiazepine nucleus, which replaces the ketone at position 2 of the nucleus. The imidazole ring allows midazolam to readily form salts, which have increased aqueous solubility and stability compared with other benzodiazepines. The exact mechanism of action for midazolam is not fully understood, but it is thought to involve potentiation of GABAergic neurotransmission resulting from binding at the benzodiazepine site of the GABA-A receptor. Midazolam is primarily metabolized by the liver and intestinal CYP3A4 to its pharmacologic active metabolite, 1-hydroxy midazolam (also termed alpha-hydroxymidazolam), which is at least as potent as the parent compound. Midazolam intranasal is indicated for acute treatment of intermittent, stereotypic episodes of frequent seizure activity (ie, seizure clusters, acute repetitive seizures) that are distinct from a patient’s usual seizure pattern in adults and children aged 12 years or older with epilepsy.
Approval was based on the ARTEMIS-1 clinical trial (n=262), Patients randomized to treatment, 201 treated a seizure cluster with double-blind study drug (n=134 midazolam intranasal; n=67 placebo) and were included in the modified intent-to-treat population. [40] The proportion of subjects with treatment success was significantly greater in those treated with midazolam intranasal (53.7%) compared with placebo (34.4%; P=.0109). The proportion of subjects with recurrence of seizure(s) from 10 minutes to 4 hours after study drug administration was significantly lower with midazolam intranasal (38.1%) compared with placebo (59.7%; P=.0043).
Diazepam intranasal
Diazepam intranasal is indicated for acute treatment of intermittent, stereotypic episodes of frequent seizure activity (ie, seizure clusters, acute repetitive seizures) that are distinct from the usual seizure pattern in patients with epilepsy who are 6 years or older.
As with other benzodiazepines, the mechanism of action is thought to potentiate GABA neurotransmission by binding to the GABA-A receptor. Diazepam is primarily metabolized by CYP3A4 and CYP2C19. Diazepam intranasal is metabolized to an active metabolite (desmethyldiazepam). The drug has a half-life of nearly 50 hours (including its active metabolite).
The dose is dependent upon the age and weight of the individual. It is administered as a single dose, with an option to repeat once after 4 hours if needed.
Efficacy for approval by the US Food and Drug Administration was based on the relative bioavailability of the nasal spray compared with that of diazepam rectal gel in healthy adults. Diazepam systemic exposure was measured by pharmacokinetic parameters (ie, peak plasma concentration [Cmax], area under the curve [AUC]). The diazepam pharmacokinetic parameters for diazepam intranasal were 2- to 4-fold less variable than those for diazepam rectal gel. [41]
In an open-label, phase 3 trial (n=121), 1585 seizure episodes were treated with diazepam intranasal in patients with frequent breakthrough seizures or acute repetitive seizures. A single dose was sufficient to achieve seizure control in 92% (1457) of seizure episodes. [42]
Phenobarbital
Phenobarbital (PHB) is the most commonly prescribed AED of the 20th century. It is a very potent anticonvulsant with a broad spectrum of action. Currently, its use is limited because of its adverse effects. It is a free acid, relatively insoluble in water. The sodium salt is soluble in water but unstable in solution. It has a direct action on GABA-A receptors by binding to the barbiturate-binding site that prolongs the duration of chloride channel opening. It also reduces sodium and potassium conductance and calcium influx and depresses glutamate excitability.
PHB is a powerful inducer of the hepatic microsomal enzymes. It has an oral or IM bioavailability of 80-100% in adults. Time to peak plasma level is 1-3 hours, but it may be delayed after oral administration in patients with poor gastrointestinal (GI) motility. The serum peak levels after IM injection are achieved in 4 hours. Ethanol increases the rate of PHB absorption. It is absorbed mainly in the small intestine.
Plasma protein binding is 40-60%. The concentration in breast milk is approximately 40% of the serum concentration. The volume of distribution ranges from 0.42-0.75 L/kg. A change in pH causes a shift of the drug between compartments; therefore, acidosis increases the concentration of PHB in the tissue compartment.
After IV administration, PHB is distributed quickly to highly vascular organs, except the brain, and then it is distributed evenly. After 6-12 minutes, it penetrates the brain, but the brain penetration is much faster during status epilepticus because of increased blood flow and acidosis.
PHB has a very long elimination half-life (ie, 75-120 h); in infants, the half-life is much longer, up to 400 hours. In individuals older than 6 months, the half-life falls to 20-75 hours. PHB is metabolized in the liver. The major metabolite is p-hydroxy phenobarbital, which is excreted as a glucuronide conjugate. PHB has extensive urinary resorption, which is enhanced by acidification of the urine.
Metabolism of PHB is inhibited by phenytoin (PHT), valproate (VPA), felbamate, and dextropropoxyphene. Enzyme inducers, such as rifampin, decrease PHB levels. Because of the potent induction of the hepatic enzymes, PHB increases the metabolism of estrogen, steroids, warfarin, carbamazepine (CBZ), diazepam, clonazepam, and VPA. Its effect on PHT is unpredictable.
In a multicenter double-blind study, PHB was found to be as effective as PHT and CBZ in the treatment of partial and secondarily generalized seizures. The Veterans Administration (VA) cooperative study, however, which compared PHB, primidone, PHT, and CBZ, showed a significantly lower retention in patients on PHB or primidone, despite their similar efficacy, because of poorer tolerability. No statistical difference was reported between PHT and CBZ.
PHB is effective in a wide variety of seizures and is currently the cheaper AED. PHB still is a first-line drug for treatment of status epilepticus. However, because of its adverse effects (eg, sedation and cognitive slowing), it is a second-line agent in the treatment of partial onset and secondarily generalized tonic-clonic seizures. In developing countries, it is used widely because of its low cost.
PHB is available in tablets of 15 mg, 30 mg, 50 mg, 60 mg, and 100 mg; elixirs (15 mg/mL); and injections (200 mg/mL). The usual starting dose is 30-60 mg once a day. The dose can be titrated up to 240 mg/d. Slow titration is better tolerated. Therapeutic blood levels are 15-40 mg/L. Physical dependence and withdrawal seizures occur with long-term use. Therefore, very slow withdrawal over several weeks to months is recommended.
The most important adverse effects of PHB are cognitive and behavior alterations. Children are more likely than adults to exhibit behavioral changes (eg, paradoxical hyperkinesis). Sedation is prominent, particularly at the beginning of therapy, and usually subsides. Psychomotor slowing, poor concentration, depression, irritability, ataxia, and decreased libido are other effects.
Long-term use of PHB may be associated with coarsening of facial features, osteomalacia, and Dupuytren contractures. Folate deficiency, megaloblastic anemia, and idiosyncratic skin reaction are rare. Vitamin supplementation is warranted. Hepatitis has been reported secondary to an immune-mediated process.
Primidone
Primidone is metabolized to PHB and phenylethylmalonamide (PEMA). Its main action is through the derived PHB. The real clinical effect of primidone or PEMA is unknown and controversial.
Primidone is absorbed orally. Bioavailability is close to 100%, with a peak level after 3 hours. Plasma protein binding is only 25%. Elimination half-life is 5-18 hours, but that of the derived PHB is 75-120 hours. Primidone is metabolized by the cytochrome oxidase system; therefore, it is affected by enzyme inducers, including PHB itself. The levels of primidone seldom are useful for monitoring efficacy, and range from 5-12 mg/L. The PHB level is the same as the level when PHB is administered directly (15-40 mg/L).
Primidone has the same indications as PHB. It is available in tablets of 50 mg and 250 mg and suspension of 250 mg/5 mL; 250 mg of primidone is equivalent to 60 mg of PHB. The average therapeutic doses range from 500-1500 mg.
The major adverse effects of primidone are intense sedation, dizziness, and nausea at the onset of treatment, most likely secondary to administration of the parental drug. These effects usually clear after 1 week of treatment. A very low dose is recommended at the onset of treatment. Other effects are the same as those of PHB.
Primidone can be used for partial onset and secondarily generalized seizures. However, it is a second-line agent because of its side-effect profile, which is similar to that of PHB. It has been useful in the treatment of essential tremor at low doses.
GABA Reuptake Inhibitors
Reuptake of gamma-aminobutyric acid (GABA) is facilitated by at least 4 specific GABA-transporting compounds; these carry GABA from the synaptic space into neurons and glial cells, where it is metabolized. Nipecotic acid and tiagabine (TGB) are inhibitors of these transporters; this inhibition makes increased amounts of GABA available in the synaptic cleft. GABA prolongs inhibitory postsynaptic potentials (IPSPs).
Tiagabine
Tiagabine (TGB) is a derivative of the GABA uptake inhibitor nipecotic acid. It acts by inhibition of the GABA transporter-1 (GAT-1). This inhibitory effect is reversible. TGB is lipid soluble and thus is able to cross the blood-brain barrier. It was introduced into clinical practice in 1998. Measurements in human and experimental models have confirmed that extracellular GABA concentrations increase after administration of TGB. Studies have shown little or no effect at other receptor systems.
The oral bioavailability of TGB is approximately 96%. The time to peak concentrations is approximately 1 hour after oral intake. A second peak of the plasma concentration of TGB is seen 12 hours after ingestion, probably caused by enterohepatic circulation. Food decreases absorption 2- or 3-fold; however, the total amount absorbed is unchanged by food administration.
TGB’s volume of distribution is 1 L/kg, and the drug is bound extensively (ie, 96%) to human plasma proteins. It is metabolized extensively in the liver by the P-450 system. None of the TGB metabolites has any antiepileptic action, and less than 3% of the drug appears unchanged in the urine.
The plasma half-life of TGB has been found to range from 4.5 to 8.1 hours in healthy volunteers, and this is reduced to 3.8-4.9 hours in patients with epilepsy who are comedicated with enzyme-inducing drugs. The clearance of TGB is greater in children. The elimination of the drug is reduced in patients with mild to moderately severe liver impairment.
TGB causes a small decrease in valproate (VPA). It has no significant effects on plasma concentrations of progesterone, estradiol, follicle-stimulating hormone, or luteinizing hormone. Hepatic-inducing drugs increase the clearance of TGB by two thirds. TGB plasma concentrations are not affected by VPA, cimetidine, or erythromycin.
TGB has been studied as adjunctive therapy in 5 double-blind, placebo-controlled studies, which demonstrated its efficacy. Besides these 5 studies, TGB has been the subject of other clinical trials designed to demonstrate efficacy, including 3 trials (1 open and 2 double-blinded) in monotherapy and 6 open long-term studies. In a meta-analysis comparing these results with placebo-controlled, randomized trials of other drugs, no significant differences in efficacy were demonstrated among TGB, gabapentin, lamotrigine (LTG), topiramate, vigabatrin (VGB), and zonisamide (ZNS).
In the long-term extension studies, 772 patients were treated with TGB (< 80 mg/d), with reduction in seizure frequency by 50% or more in about 30-40% of patients treated for 3-6 months. This effect was maintained for 12 months in patients with partial seizures but not in patients with secondarily generalized seizures. The drug is available for use as second-line add-on therapy in patients with partial or secondarily generalized seizures that are refractory to treatment.
In the United States, the recommended dosage is 4 mg/d with a titration of 4-8 mg/d each week, and the usual maintenance dose is 32-56 mg/d. In Europe, the recommended dose is 15 mg/d followed by weekly incremental increases of 5-15 mg up to a maximum of 15-30 mg/d. In patients on concomitant enzyme inducers, the dose could be increased gradually to 45 mg/d.
The most troublesome adverse effects of TGB include dizziness, asthenia, nervousness, tremor, depressed mood, and emotional lability. Diarrhea also is significantly more frequent among TGB-treated patients than placebo-treated patients. Other adverse effects include somnolence, headaches, abnormal thinking, abdominal pain, pharyngitis, ataxia, confusion, psychosis, and skin rash. No changes in biochemical or hematologic parameters are reported. Serious idiosyncratic adverse effects are recorded as commonly in patients on placebo as in those on TGB.
A few clinical trials have reported the occurrence of convulsive and nonconvulsive status epilepticus with TGB. In the author’s experience, 1 case of nonconvulsive status was caused by accidental overdose. TGB therapy should be used cautiously in patients with a history of status epilepticus. TGB is contraindicated in severe hepatic impairment, pregnancy, and lactation.
Use of TGB is limited to adjunctive therapy in refractory partial epilepsy. It should not be used in absence epilepsy or in partial epilepsies with generalized spike wave, since it can worsen seizure control or cause status epilepticus.
GABA Transaminase Inhibitors
Gamma-aminobutyric acid (GABA) is metabolized by transamination in the extracellular compartment by GABA-transaminase (GABA-T). Inhibition of this enzymatic process leads to an increase in the extracellular concentration of GABA. Vigabatrin inhibits the enzyme GABA-T.
Vigabatrin
In the 1970s, GABA was recognized as an important inhibitory neurotransmitter in the central nervous system (CNS). Favoring the balance toward the GABA system was a major target of drug research, and soon vigabatrin (VGB) was developed. The US Food and Drug Administration approved VGB in 2009. Although effective, it can cause vision loss, and because of this risk, it is not used as a first-line agent and is available only through a restricted access program.
VGB is indicated as adjunctive therapy for adults and pediatric patients aged 2 years or older with refractory complex partial seizures who have inadequately responded to several alternative treatments and for whom the potential benefits outweigh the risk of vision loss.
Additionally, it is indicated as monotherapy for infantile spasms in pediatric patients aged 1 month to 2 years for whom the potential benefits outweigh the potential risk of vision loss.
VGB is a close structural analogue of GABA, binding irreversibly to the active site of GABA-T. Newly synthesized enzymes take 4-6 days to normalize the enzymatic activity. In vivo studies in human and animal subjects have shown that VGB significantly increases extracellular GABA concentrations in the brain. VGB has no other known action. [43, 44, 45, 46]
VGB is highly soluble in water but only slightly soluble in ethanol. It is absorbed rapidly after oral ingestion, with an oral bioavailability of 100%. Time to peak concentration is approximately 2 hours, and the volume of distribution of the drug is 0.8 L/kg. About 10% of the plasma concentration is found in cerebrospinal fluid (CSF). Only a small fraction crosses the placenta.
VGB is excreted in urine (up to 95%) with a half-life of 4-7 hours. In elderly patients, clearance is reduced and the half-life may double. VGB does not induce the activity of hepatic enzymes. Correlation between plasma levels and clinical effect is poor.
VGB can reduce the plasma concentration of phenytoin (PHT) by 25%. This reduction probably is mediated by decreased absorption; however, the exact mechanism is unknown. No other pharmacokinetic or pharmacodynamic interactions are present.
VGB has been studied exhaustively in 9 double-blind controlled trials. These trials reported that 40-50% patients with refractory partial seizures had a reduction in seizure frequency of more than 50%, and as many as 10% of patients became seizure free. Many patients continued the drug after completion of the trial. The dose of VGB ranged from 1000-4000 mg/d.
VGB is less effective against primarily generalized tonic-clonic seizures and also may worsen myoclonic seizures or generalized absence seizures. Like tiagabine (TGB), VGB has been reported to cause absence status. Patients with myoclonus or Lennox-Gastaut syndrome do not respond well to VGB.
In placebo-controlled trials in patients with refractory epilepsy, 20% of children and 5% of adults showed an increase in seizures. VGB is very effective in the treatment of infantile spasms; therefore, it is the drug of choice for this indication in many countries.
The usual starting dose for adults is 500 mg twice daily, and this is increased by 250-500 mg every 1-2 weeks to a maximum dose of 4000 mg/d. In children, 40 mg/kg/d is the usual starting dose, with maintenance doses of 80-100 mg/kg.
The most common adverse effect of VGB is drowsiness. Other important adverse effects include neuropsychiatric symptoms, such as depression (5%), agitation (7%), confusion and, rarely, psychosis. Minor adverse effects, usually occurring at the onset of therapy, include fatigue, headache, dizziness, increase in weight, tremor, double vision, and abnormal vision. VGB has little effect on cognitive function. Acute hypersensitivity and idiosyncratic immunologic adverse effects are extremely rare.
VGB causes widespread intramyelinic vacuolization throughout the brains of rats and dogs; however, primate and human studies have not demonstrated such changes. VGB also affects the retina in some rodent species, and later human studies show visual field changes, characterized by nasal constriction and then concentric constriction, with preservation of central vision. In 1 series, visual field disturbances were found in more than 50% of cases. The mechanism of this effect is unknown and the risk factors are unclear. In some cases, it appears to be irreversible.
AEDs with Potential GABA Mechanism of Action
The enzyme glutamic acid decarboxylase (GAD) converts glutamate into gamma-aminobutyric acid (GABA). Currently, valproate (VPA) and gabapentin (GBP) are known to have some effect on this enzyme and thereby enhance the synthesis of GABA, in addition to other potential mechanisms of action. VPA also blocks the neuronal sodium channel during rapid sustained repetitive firing. GBP has a weak competitive inhibition of the enzyme GABA-T.
As with other antiepileptic drugs (AEDs), whether these mechanisms of action alone are responsible for the antiseizure efficacy of VPA and GBP is unclear.
Gabapentin
Gabapentin (GBP) was developed to have a structure similar to that of GABA; however, experimental evidence showed that GBP has, in fact, little or no action on the GABA receptor. It is highly soluble in water. It enhances GAD but does so weakly. It binds with the alpha2 delta subunit of calcium channels in the cerebral neocortex, hippocampus, and spinal cord; this mechanism of action may be important for its efficacy in pain.
At this time, the exact mechanism by which GBP increases the intracellular concentration of GABA is unknown. In vivo magnetic resonance spectroscopy studies have shown that GBP increases brain levels of GABA and its metabolites homocarnosine and pyrrolidinone. It also may reduce monoamines and affect serotonin release.
GBP is a competitive inhibitor of the enzyme branched chain amino acid transferase, which metabolizes the branched-chain amino acids (leucine, isoleucine, and valine) to glutamate. Through this mechanism, GBP may reduce brain glutamate levels. [47, 48, 49]
GBP has a bioavailability of less than 60%; bioavailability is affected mainly by variable absorption, which depends on an L-amino acid transporter. Absorption may be impaired in some clinical situations in which active transport usually is compromised. In addition, single doses of GBP greater than 1200 mg decrease the bioavailability to 35%.
Once absorbed, the drug readily crosses the blood-brain barrier and achieves a plasma-to-CSF ratio of approximately 1:10. Peak serum levels are achieved within 2-4 hours of oral administration. The volume of distribution in adults is about 0.64-1.04 L/kg at steady state.
GBP is not bound to plasma proteins and is not metabolized. It does not induce hepatic enzymes. It is excreted entirely in an unchanged form. The renal clearance of 120-130 mL/min is correlated linearly with creatinine clearance. The elimination half-life of the drug is 5-9 hours.
Steady-state levels are achieved within a few days, and the half-life does not change with chronic administration, nor is it influenced by concomitant medications. Few data on the correlation between serum level and effectiveness are reported. In patients with renal disorders, the dose should be adjusted according to creatinine clearance; it is removed during hemodialysis.
GBP has no pharmacokinetic drug interactions. However, antacids can reduce the bioavailability of GBP.
Several open and double-blind trials have been conducted with GBP. In the United States, patients were randomized to receive 600 mg, 1200 mg, or 1800 mg of GBP or placebo; the percentage of patients who had a reduction of seizures of 50% or more was 18-26% with GBP and 8% with placebo. A large multicenter study carried out in the United Kingdom randomized patients to receive add-on therapy with either GBP 1200 mg or placebo and showed a reduction in partial seizures of 50% or more in 28% of patients taking GBP and 9.8% of patients taking placebo.
A double-blind study in children with partial epilepsy showed response rates of 17% on GBP and 7% on placebo, whereas in other double-blind, placebo-controlled studies, GBP had no effect in childhood absence seizures. These trials were performed at a relatively low dose, and a better response was obtained in trials using a higher dose; however, the latest trials were not double blind.
In clinical practice, higher doses often are used. GBP is useful in the treatment of partial and secondarily generalized tonic-clonic seizures but is ineffective in myoclonus and in most generalized seizure disorders. The drug appears to have only a modest efficacy, particularly at lower doses.
GBP is available as capsules of 100 mg, 300 mg, 400 mg, and 600 mg and tablets of 800 mg. Rapid titration is well tolerated in some patients, but usually the drug is titrated at weekly intervals to a maximum of 3600-4800 mg/d.
Its lack of drug interactions, lack of plasma protein binding, and renal excretion make GBP particularly useful in patients with renal or hepatic disease and in patients on complex drug regimens. Patients with coexistent migraine headache or neuropathic pain may benefit from this drug.
GBP is relatively well tolerated; it does have some adverse effects, particularly in high doses, but these usually are relatively minor. No significant serious idiosyncratic or systemic adverse effects have been reported. The incidence of rash is 0.5% and of neutropenia, 0.2%. Electroencephalographic (EEG) changes and/or angina were found in 0.05%. No cases of hepatotoxicity have been recorded.
In the early double-blind studies, 44% of patients reported adverse effects with 900 mg of GBP. Similar adverse effects were recorded in later studies with 1200 mg. In a US study of patients taking 1800 mg/d, somnolence was recorded in 36% of patients, dizziness in 24%, ataxia in 26%, nystagmus in 17%, headache in 9%, tremor in 15%, fatigue in 11%, diplopia in 11%, rhinitis in 11%, and nausea or vomiting in 6%. Most of these effects were mild.
Pregabalin
Pregabalin is an analogue of the neurotransmitter GABA and has analgesic, anticonvulsant, and anxiolytic effects. [50, 51] Despite being a GABA analogue, pregabalin is inactive at GABA receptors, including GABA-A, benzodiazepine, t-butylbicyclophosphorothionate (TBPS), and GABA-B radioligand binding sites. [52] Neither pregabalin nor GBP alters GABA concentration in brain tissues [53] or inhibits GABA transport in vitro. Pregabalin binds with high affinity to both the alpha2 delta-1 and alpha2 delta-2 subtypes. [54]
GBP and pregabalin binding to the alpha2 delta protein are proposed to mediate the functional effects these molecules have on calcium currents in activated neurons and on stimulated neurotransmitter release. [55] The effect is a reduced release of excitatory neurotransmitters and peptide neuromodulators under membrane hyperexcitability, which is postulated to mediate the analgesic, anxiolytic, and anticonvulsant effect. [56]
Pregabalin is active in several animal models of seizures. In the high-intensity electroshock test, pregabalin inhibited tonic extensor seizures in rats and low-intensity electroshock seizures in mice. In the DBA/2 audiogenic mouse model and clonic seizures, pregabalin prevented tonic extensor seizures from pentylenetetrazole in mice. In a kindled rat model of partial seizures, pregabalin prevented stages 4–5 behavioral seizures. However, pregabalin was not effective in models of absence seizures. [57]
Pregabalin is well absorbed after oral administration. When the drug has been given orally under fasting conditions, the peak plasma concentration is 1.5 hours; however, when it is given with food, the rate of pregabalin absorption is decreased, resulting in a decrease in peak plasma concentration of approximately 25-30% and an increase in time to peak plasma concentration (Tmax) to approximately 3 hours. Oral bioavailability is 90% and it is independent of dose and presence of food. Steady state is achieved within 24-48 hours.
Pregabalin does not bind to plasma proteins. The apparent volume of distribution after oral administration is approximately 0.5 L/kg. Pregabalin crosses the blood-brain barrier in animals and has been shown to cross the placenta in rats and is present in the milk of lactating rats.
After oral administration, approximately 90% is recovered in the urine as unchanged pregabalin. Only about 0.9% is found in urine as the N -methylated derivative.
Pregabalin is eliminated primarily by renal excretion, with a mean elimination half-life of 6.3 hours in patients with normal renal function. Pregabalin elimination is nearly proportional to creatinine clearance (CrCl). Dosage reduction in patients with renal failure is necessary. Pregabalin is effectively removed from plasma by hemodialysis (50% after 4 h of hemodialysis).
Pregabalin has no pharmacokinetic drug interactions.
Pregabalin is indicated as adjunctive therapy for partial-onset seizures in adults and children aged 1 month or older. Efficacy as add-on therapy for partial epilepsy has been demonstrated in four major trials. [58, 59, 60, 61] Patients given pregabalin were significantly more likely to achieve a 50% or greater reduction in seizure frequency. Subgroup analyses assessing the effect of individual doses of 50 mg pregabalin was not effective. [62]
Pregabalin’s lack of drug interactions, lack of plasma protein binding, and renal excretion make it particularly useful in patients with renal or hepatic disease and in patients on complex drug regimens. Patients with coexisting migraine headache or neuropathic pain may benefit from this drug.
Pregabalin is relatively well tolerated, although it does have some adverse effects, particularly in high doses. No significant serious idiosyncratic or systemic adverse effects have been reported.
The most common side effects of pregabalin are dizziness and drowsiness. Other important side effects include dry mouth, edema, blurred vision, weight gain, and difficulty concentrating. Pregabalin has rarely been associated with angioedema (swelling of the face, tongue, lips, and gums, throat, and larynx).
Valproate
Valporate (VPA) is one of the most commonly used AEDs around the world. It is the drug of choice for primary generalized epilepsies and is also approved for the treatment of partial seizures. It was discovered by accident; first synthesized in 1882, its antiepileptic properties were recognized when it was used as a solvent for the experimental screening of new AEDs. It was licensed in Europe in the early 1960s, where its use became extensive. It has been used in different forms (eg, divalproex sodium, magnesium or calcium salt, and valpromide), which do not differ significantly.
The mechanism of action is uncertain. VPA enhances GABA function, but this effect is observed only at high concentrations. It may increase the synthesis of GABA by stimulating GAD. It also produces selective modulation of voltage-gated sodium currents during sustained, rapid, repetitive neuronal firing. [63, 64, 65]
VPA is a simple molecule, similar to endogenous fatty acids. It is slightly soluble in water and highly soluble in organic solvents. The sodium salt is highly water soluble, whereas the calcium and magnesium salts are insoluble. VPA is absorbed rapidly and nearly completely, with a bioavailability close to 100%.
The peak plasma level after oral administration is reached in 13 minutes to 2 hours. The acid form takes a longer time to reach peak plasma concentration (3-4 h), and divalproex sodium reaches peak plasma concentration slightly faster. The enteric-coated divalproex sodium reaches peak concentration in approximately 3-8 hours. The sprinkle form peaks in 4 hours.
VPA is 85-95% bound to plasma proteins. Protein binding decreases at higher levels (ie, >100 mg/mL), in renal and hepatic diseases, and during pregnancy. Some other drugs (eg, aspirin, phenylbutazone) displace VPA, but other AEDs do not. Total serum VPA levels affect protein binding so that at serum concentrations lower than 75 µg/mL, the unbound fraction ranges from 7% to 9%. At serum concentrations of 100 µg /mL, the free fraction increases to 15%. The volume of distribution is 0.1-0.4 L/kg.
VPA reaches the brain by an active transport process that is saturable. It is metabolized in the liver (96%) mainly by beta-oxidation and then glucuronidation. Less than 4% is excreted unchanged in urine. Some metabolites may be responsible for adverse effects, particularly the 4-ene metabolite, which may cause hepatic toxicity. The plasma half-life is 16 hours. When VPA is used with enzyme-inducing AEDs, the half-life is reduced to 9 hours.
An intravenous (IV) form of VPA (Depacon; Abbott Pharmaceuticals) is available. It is well tolerated, with only 2% of patients discontinuing treatment because of adverse effects, and has no significant effect on the cardiovascular system. Bioavailability is similar to that of the oral preparation. The Tmax after IV VPA is at the end of 1-hour infusion, and thus the drug should be administered every 6 hours.
VPA is a potent inhibitor of both oxidation and glucuronidation. It increases plasma levels of free fractions of phenytoin (PHT), phenobarbital (PHB), carbamazepine (CBZ) epoxide, and lamotrigine (LTG). It reduces the total PHT level. The levels of VPA are decreased by enzyme-inducing drugs and are increased by felbamate and clobazam.
VPA is a potent AED, effective against a wide range of seizure types. It is the drug of choice in idiopathic generalized epilepsy. Open and comparative studies have shown excellent control rates in patients with newly diagnosed typical absence seizure. It is the drug of choice for juvenile myoclonic epilepsy and can be used in other types of myoclonus. In addition, it is a first-line drug in photosensitive epilepsy and Lennox-Gastaut syndrome. It is a second choice in the treatment of infantile spasms. In focal epilepsy, VPA has been shown to be as effective as other first-line agents.
The usual starting dose is 250 mg/d, with a maintenance dose of 500-1500 mg/d. In the author’s opinion, 3 times–daily dosing is better tolerated, but twice-daily dosage is usual. Rapid titration usually is well tolerated. In children, the usual dose is 20 mg/kg/d and the maintenance dose is 40 mg/kg. Serum level has poor correlation with clinical effect and has significant daily fluctuations. IV VPA should be administered as a 60-minute infusion with a rate not exceeding 20 mg/min.
VPA is available as 125 mg, 250 mg, and 500 mg delayed-release tablets; 125 mg and 250 mg sprinkle capsules; 500 mg extended-release tablets; 250 mg/5 mL syrup; and parenteral preparation for IV injection.
Meador et al found that in utero exposure to VPA, as compared with other antiepileptic agents, is associated with a lower IQ in children. [66] The study took place over 5 years in 25 epilepsy centers in the United States and the United Kingdom. The design was a prospective, observational, cohort study of pregnant women with epilepsy who took a single agent (CBZ, LTG, PHT, or PHB).
The cohort study assessed the neurodevelopmental outcomes of children who were exposed in utero to several antiepileptic drugs. A planned interim analysis conducted when the children were 3 years of age found an increased risk of impaired cognitive function compared with other commonly used antiepileptic drugs; this association was dose-dependent. The investigators concluded that VPA should not be used as a first-line agent in women of childbearing potential. [66]
This interim analysis was updated in 2013 with data from children completing 6 years of follow-up. The final results of the Neurodevelopmental Effects of Antiepileptic Drugs (NEAD) study showed that children exposed to valproate products while their mothers were pregnant had decreased IQs at age 6 compared to children exposed to other antiepileptic drugs. [67]
The prescribing information was changed to reflect this risk including a Black Box Warning stating not to use valproate derivatives in women of childbearing age unless the drug is essential to the management of seizures, or for manic episodes associated with bipolar disorder (FDA fetal risk category D). It should not be used for migraine headache prophylaxis during pregnancy (FDA fetal risk category X).
Even though VPA has been used for many years, large controlled and blinded studies to determine the frequency of adverse effects have not been conducted. On the basis of clinical experience, dose-related adverse effects include nausea, vomiting (mainly during initiation of therapy and improved by administration of enteric-coated preparations), tremor, sedation, confusion or irritability, and weight gain.
Metabolic effects from interference in mitochondrial metabolism include hypocarnitinemia, hyperglycinemia, and hyperammonemia. Severe sedation or even coma may result from hyperammonemia, typically with normal liver function tests. Patients with an underlying urea cycle enzyme defect may become encephalopathic from acute hyperammonemia, which may be fatal occasionally.
Hair loss or curling of hair may occur, which in the author’s patients has improved when the patients used baby shampoo and a multivitamin supplement. VPA has adverse endocrine effects, including insulin resistance and change in sex hormone levels causing anovulatory cycles, amenorrhea, and polycystic ovary syndrome. Bone marrow suppression with neutropenia and allergic rashes are rare. Acute pancreatitis is rare but potentially fatal and usually reverses after withdrawal of VPA.
The most serious idiosyncratic adverse effect is hepatotoxicity. This is observed mainly in patients younger than 2 years and with polytherapy.
Because of its adverse-effect profile, VPA is being replaced by newer AEDs. The new extended-release preparation may decrease dose-related adverse effects and may be better tolerated. It may be useful in patients with concomitant migraine headache. Hepatic failure from VPA is extremely rare in adulthood. VPA should be used with caution in women of reproductive age.
The inhibition of kindling in experimental models suggests a potential use as a prophylactic agent for seizures; however, no clinical studies support this hypothesis. IV VPA is useful in patients when oral administration is not possible and when rapid IV loading is necessary. It is also helpful in patients with poorly controlled repetitive seizures that require a rapid IV load. Unlike PHT or PHB, IV VPA is not associated with hemodynamic changes.
Ganaxolone
Ganaxolone is a GABAA receptor positive modulator. It binds specifically to GABAA receptors to enhance their inhibitory effects. It is indicated for seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) in patients aged 2 years or older. CDD, a rare developmental epileptic encephalopathy, is largely a disease of pediatric and young adult patients.
Approval was based on the phase 3 MARIGOLD trial. Patients treated with ganaxolone (n = 49) showed a median 30.7% reduction in 28-day major motor seizure frequency, compared with a 6.9% reduction for those receiving placebo (n = 51) (P = .0036). In the open label extension study, patients treated with ganaxolone for at least 12 months (n = 48) experienced a median 49.6% reduction in major motor seizure frequency. [68]
Glutamate Blockers
Glutamate and aspartate are the most two important excitatory neurotransmitters in the brain. The glutamate system is a complex system that contains macromolecular receptors with different binding sites (ie, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid [AMPA], kainate, N -methyl-D-aspartate [NMDA], glycine, and metabotropic sites).
The AMPA and the kainate sites open a channel through the receptor, allowing sodium and small amounts of calcium to enter. The NMDA site opens a channel that allows large amounts of calcium to enter along with the sodium ions. This channel is blocked by magnesium in the resting state. The glycine site facilitates the opening of the NMDA receptor channel. The metabotropic site is regulated by complex reactions and its response is mediated by second messengers.
NMDA antagonists have a limited use because they produce psychosis and hallucinations. In addition to these adverse effects, learning and memory may be impaired by blocking these receptors, because NMDA receptors are associated with learning processes and long-term potentiation.
Felbamate
Felbamate is a potent anticonvulsant, very effective against multiple seizure types. Unfortunately, after the occurrence of aplastic anemia and hepatic failure, approval for general use was withdrawn. It is now available in the United States only for a very limited use, principally by neurologists in patients for whom potential benefit outweighs the risk. It blocks the NMDA receptors and voltage-gated calcium channels and also modulates sodium-channel conductance, but has no effect on gamma-aminobutyric acid (GABA) receptors. [69, 70, 71]
In addition to its activity against seizures, felbamate has been shown to have a neuroprotective effect on models of hypoxic-ischemic injuries. Wallis and Panizzon reported neuroprotection after treatment with felbamate in the rat hippocampal slice model following hypoxic exposure. [72] Wasterlain et al also demonstrated the neuroprotective effect of felbamate after bilateral carotid ligation in rat pups with a window of opportunity for neuroprotection of 1-4 hours after the ligation. [73]
After oral administration, felbamate is well absorbed, with a time to peak plasma concentration Tmax) of 1-4 hours after the dose. It is distributed rapidly throughout the body, including the brain. Lipid-mediated blood-brain barrier penetration of felbamate is similar to that of phenytoin (PHT) and phenobarbital (PHB). It is 20-25% bound to plasma proteins (mainly albumin).
The liver, via hydroxylation and conjugation, extensively metabolizes felbamate. The major identified metabolites of felbamate are p-hydroxy-felbamate, 2-hydroxy-felbamate, a monocarbonate, and a 3-carbamoyl oxy-2-phenylpropionic acid. Approximately 40-49% of a felbamate dose is recovered in the urine as the parent compound. The elimination half-life of felbamate ranges from 13 to 30 hours when the drug is administered as monotherapy.
Concomitant enzyme inducers decrease the half-life to 13-14 hours, and the concentration of felbamate is significantly higher than expected in the presence of valproate (VPA). Felbamate also increases PHT levels and reduces carbamazepine (CBZ) levels with increments in CBZ-epoxide levels.
Because of its potentially fatal toxic effects (in particular, the small but definitive risk of aplastic anemia and hepatic failure), use of felbamate is restricted to patients with severe partial epilepsy or Lennox-Gastaut syndrome who do not respond to other medications. The reported therapeutic serum level ranges from 30-100 mg/L. Initial dose of 1200 mg/d in 3 or 4 divided doses, with titration of 600 mg/wk up to a maximum of 2400-3600 mg/d, is recommended. Reducing the dosage of concomitant antiepileptic drugs (AEDs) can eliminate most adverse effects.
In children, recommended starting dose is 15 mg/kg/d, with weekly increments as high as 45 mg/kg/d. Again, concomitant AEDs should be reduced by 20% or more upon initiation of treatment and reduced further on the basis of symptoms and blood levels. Felbamate is available as 400 mg and 600 mg tablets and 600 mg/5 mL suspension.
Felbamate usually is well tolerated. Common adverse effects include insomnia, weight loss, nausea, decreased appetite, dizziness, fatigue, ataxia, and lethargy. Polytherapy is associated with increases in adverse effects. Clinical trials have shown that approximately 12% of patients discontinue the drug because of adverse effects. Fatal hepatic failure has been reported in 14 of 110,000 treated patients. Besides polytherapy, no other risk factor has been found. Most of the deaths occurred within 6 months of initiation of therapy.
Topiramate
Topiramate is a very potent anticonvulsant that is structurally different from other AEDs. It is derived from D-fructose and initially was developed as an antidiabetic drug. In animal models, it was found to have potent antiepileptic effects. Topiramate has multiple mechanisms of action. It exerts an inhibitory effect on sodium conductance, decreasing the duration of spontaneous bursts and the frequency of generated action potentials, enhances GABA by unknown mechanisms, inhibits the AMPA subtype glutamate receptor, and is a weak inhibitor of carbonic anhydrase.
Topiramate is absorbed rapidly after oral administration and has a bioavailability close to 100%. When it is administered at regular doses, food delays but does not affect the extent of absorption. The time to peak blood levels is about 2 hours. The volume of distribution ranges from 0.6-1.0 L/kg. Plasma protein binding is approximately 15%.
Only 15% of topiramate is metabolized in the liver by the P-450 microsomal enzyme system. None of the metabolites has antiepileptic action, and the majority of the drug (ie, 85%) is excreted unchanged in the urine. However, metabolism is much more extensive in patients on polytherapy, presumably as a result of enzyme induction.
In patients with renal failure, doses may have to be reduced. The elimination half-life ranges from 18-23 hours and is independent of dose over the normal clinical range. In experimental settings, no tolerance to topiramate has been recorded.
Enzyme-inducing drugs, such as PHT and CBZ, decrease serum topiramate concentrations by approximately 50%. Topiramate generally does not affect the steady-state concentrations of the other drugs given in polytherapy, although PHT levels may rise occasionally. Topiramate reduces ethyl estradiol levels by 30% and may inactivate the low-dose contraceptive pill. It may cause a mild reduction in digoxin levels.
Topiramate has a marked antiepileptic effect, as demonstrated in 6 double-blind, parallel-group, placebo-controlled, add-on trials and in a variety of open studies. As many as 5% of the patients became seizure free in some trials, 44% had a greater than 50% reduction in seizure frequency (compared to 12% of patients on placebo), and 21% had a greater than 75% reduction in seizure frequency.
Meta-analysis of placebo-controlled parallel-group studies of topiramate and the other new AEDs has shown greater effects from topiramate than from any of the other drugs in comparison with placebo. Topiramate has also been effective as adjunctive therapy in drug-resistant generalized epilepsies, including juvenile myoclonic epilepsy, absence and generalized tonic-clonic seizures, and Lennox-Gastaut syndrome.
In the United States, topiramate currently is approved for (1) partial onset and secondarily generalized tonic-clonic seizures, (2) primary generalized tonic-clonic seizures, and (3) Lennox Gastaut syndrome.
Topiramate is available as 25 mg, 50 mg, 100 mg, and 200 mg tablets and as 15 mg and 25 mg sprinkle formulations. A parenteral form is not available.
Topiramate should be started at a low dosage and titrated slowly to prevent adverse effects. Recommended starting dosage is 25 mg/d; this is increased in weekly or biweekly increments of 25-50 mg. Maintenance dosage is 200-600 mg/d in 2 divided doses. In children, the usual starting dosage is 0.5-1 mg/kg/d with increments of 0.5-1 mg/kg/d every 2 weeks. In clinical trials, pediatric dosing in the range of 9-11 mg/kg/d produced optimal seizure control.
The most common adverse effects of topiramate include ataxia, impairment of concentration, confusion, dizziness, fatigue, paresthesia in the extremities, somnolence, disturbance of memory, depression, agitation, and slowness of speech. If the drug is continued, many adverse effects subside within a few weeks.
The most common adverse effects in children are somnolence, anorexia, fatigue, and nervousness. The drug causes weight loss in many patients, sometimes more than 10 kg, an effect that may lead to discontinuance. The weight loss appears to be related to appetite suppression. As a carbonic anhydrase inhibitor, topiramate also has a propensity to cause renal calculi; therefore, patients should be encouraged to drink plenty of fluids.
No idiosyncratic severe reactions or allergic rashes have been reported; however, in the author’s experience, several cases of pruritus have occurred, always associated with previous history of allergic rash to other medications. No hepatotoxicity, hematologic toxicity, serious gastrointestinal (GI) toxicity, or cardiotoxicity have been documented. Recently, acute myopia with angle-closure glaucoma has been reported as a rare adverse event associated with topiramate.
Most physicians agree that topiramate is a highly effective AED. The adverse cognitive effects occur more frequently at higher doses and with a rapid titration rate. These cognitive adverse effects can be reduced by using a slow titration rate of 25 mg every week, until 200 mg/d is reached. Subsequently the dose can be increased in weekly increments of 25-50 mg/d to a target dose of 400-600 mg/d. In the author’s experience, very slow titration, occasionally by increments of 25 mg/d biweekly, has improved tolerability in sensitive patients.
Obese patients with epilepsy may benefit from this drug because of its weight-loss–inducing effect. Topiramate is also indicated as a prophylactic agent in patients with migraine headaches. [74, 75, 76, 77, 78, 79, 80]
Perampanel
In October 2012, the US Food and Drug Administration (FDA) approved perampanel (Fycompa) as adjunctive treatment for partial-onset seizures (with or without secondary generalized seizures) and in June 2015 for primary generalized tonic-clonic seizures in adults or children aged 12 years or older. Perampanel is a noncompetitive antagonist of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA).
The approval for partial-onset seizures was based on 3 randomized, double-blind, placebo-controlled, multicenter trials in 1,480 patients who were not adequately controlled with 1 to 3 concomitant AEDs. A dose-ranging study by Krauss et al showed statistically significant declines in partial-onset seizure frequencies for the perampanel groups compared with placebo. Mean percentage declines for perampanel were 13.6% for 2 mg/day (P = .4197), 23.3% for 4 mg/day (P = .0026), 30.8% for 8 mg/day (P< .0001), and a 10.7% decrease for placebo. [81] Additional studies of higher perampanel doses (ie, 8 mg/day and 12 mg/day) improved seizure control in patients with uncontrolled partial-onset seizures. [82, 83]
Approval for PGTC seizures was based on a Phase 3, randomized, double-blind, placebo-controlled clinical trial (n = 162) in patients taking up to 3 antiepileptic drugs. Patients treated with perampanel (n=81) achieved a 76% median reduction in PGTC seizure frequency, which was statistically significant compared to 38% with placebo (n=81). Additionally, 64% of patients in the treatment arm experienced a >50% reduction in PGTC seizure frequency compared with placebo which attained a 40% reduction. [84]
Perampanel was assigned a DEA schedule III. The prescribing information includes a boxed warning that describes serious or life-threatening psychiatric and behavioral adverse reactions, including aggression, hostility, irritability, anger, and homicidal ideation and threats. [85] Common adverse effects include dizziness (up to 43%), somnolence, headache, fatigue, and irritability.
In the absence of other AEDs that induce CYP enzymes, the initial dose is 2 mg PO at bedtime. After 1 week, the dose may be increased by at least weekly intervals to 4-8 mg/day (titrate more slowly, by at least 2-week intervals in elderly persons). The target dosage range is 8-12 mg/day for partial onset seizures and 8 mg/day for tonic-clonic seizures (may increase to 12 mg/day if needed and tolerated).
If administered with AEDs that induce CYP enzymes (or other strong CYP inducers), an increased dose may be required (not to exceed 12 mg/day).
If hepatic impairment is evident, the daily dose should not exceed 6 mg/day or 4 mg/day for mild or moderate hepatic insufficiency, respectively. Perampanel is not recommended for use with severe hepatic or renal impairment or with hemodialysis.
AEDs with Other Mechanisms of Action
Levetiracetam
Levetiracetam (LEV) is a piracetam (S-enantiomer pyrrolidone) derivative. It was developed in the 1980s to enhance cognitive functions and for anxiolysis. [8, 86, 87, 88, 89, 90, 91, 92]
LEV is a unique antiepileptic drug (AED) in that it is ineffective in classic seizure models that screen potential compounds for antiseizure efficacy such as maximal electroshock and pentylenetetrazol in rats and mice. During preclinical evaluations, it was found to be effective in several models of seizures, including tonic and clonic audiogenic seizures in mice, tonic seizures in the maximum electroshock-seizure test in mice, and tonic seizures induced in rodents by chemoconvulsants.
Interestingly, LEV inhibits the development of pentylenetetrazol-induced amygdala kindling in mice, a situation in which other drugs such as phenytoin (PHT) and carbamazepine (CBZ) are inactive. The mechanism of action is possibly related to a brain-specific stereo-selective binding site, synaptic vesicle protein 2A (SV2A). SV2A appears to be important for the availability of calcium-dependent neurotransmitter vesicles ready to release their content. [4]
The lack of SV2A results in decreased action potential-dependent neurotransmission, while action potential-independent neurotransmission remains normal. [5, 6] In addition, it reduces bicuculline-induced hyperexcitability in rat hippocampal CA3 neurons, suggesting a mechanism that does not involve release of gamma-aminobutyric acid (GABA). LEV inhibits Ca2+ release from the inositol-trisphosphate (IP3)-sensitive stores without reducing Ca2+ storage, which could explain some of LEV’s antiepileptic properties.
LEV is absorbed rapidly after oral administration, with an oral bioavailability of approximately 100%. The peak concentration is reached approximately 0.6-1.3 hours after ingestion, and the rate of absorption is slowed by food. LEV does not bind to plasma proteins. The volume of distribution is approximately 0.5-0.7 L/kg.
LEV is minimally metabolized (approximately 27%), mainly by hydrolysis of the acetamide group. It does not involve the enzymes of the cytochrome P-450 system. The clinical relevance of the metabolites is likely to be negligible.
Approximately 66% is excreted unchanged in urine. LEV is cleared 48 hours after oral administration by glomerular filtration with partial tubular reabsorption. Although its elimination half-life is only 6-8 hours, its pharmacodynamic half-life is likely to be longer. In patients with renal insufficiency, the half-life may be increased up to 24 hours. The drug is removed during hemodialysis. LEV crosses the placenta, and fetal concentrations are similar to maternal levels.
No significant drug interactions have been identified. LEV does not inhibit cytochrome P-450 (CYP450) isoenzymes, epoxide hydrolase, or uridine diphosphate (UDP)-glucuronidation.
A multicenter, double-blind, responder-selected study evaluating LEV as monotherapy in patients with refractory partial epilepsy showed a 73.8% reduction of seizure frequency in 59.2% of the patients, and 18% of the monotherapy group remained seizure free.
Tolerability studies have demonstrated that LEV is very well tolerated. In the electroencephalographic (EEG) model, this drug induced a decrease in the number of frequent epileptiform discharges in most patients. An open-label study reported that 6 of 9 patients with juvenile myoclonic epilepsy refractory to valproate (VPA) or lamotrigine (LTG) became seizure free on LEV.
These results suggest that LEV might have a significant effect in generalized epilepsies. In March 2007, it was approved by the US Food and Drug Administration (FDA) as adjunctive treatment for primary generalized tonic-clonic seizures in adults and children aged 6 years and older.
The starting adult dosage is 500 mg twice a day, with weekly increments of 500-1000 mg/d to 3000 mg, if required. A slower titration rate of 250 mg twice a day with subsequent weekly dose increases by 500 mg is tolerated better.
Pediatric dosages are determined by seizure type and patient age. Use oral solution if weight is ≤20 kg. Pediatric recommendations for tonic-clonic seizures are as follows:
-
Younger than 6 years - Not established
-
Aged 6-15 years - 20 mg/kg/d PO divided bid; may increase daily dose by 20 mg/kg increments q2wk to recommended dose of 30 mg/kg bid
-
Aged 16 years and older - Administer as in adults
Pediatric recommendations for partial-onset seizures are as follows:
-
Younger than 1 month - Not established
-
1-5 months: 14 mg/kg/d PO divided bid; may increase by 14 mg/kg/day increments q2wk to recommended dose of 21 mg/kg bid
-
6 months through 3 years: 20 mg/kg/d PO divided bid: increase by 20 mg/kg/d increments q2wk to recommended dose of 25 mg/kg bid
-
Aged 4-15 years - 20 mg/kg/d PO divided bid; may increase by 20 mg/kg/d increments q2wk to recommended dose of 30 mg/kg bid
-
Aged 16 years and older - Administer as in adults
Pediatric recommendations for myoclonic seizures are as follows:
-
Younger than 12 years - Not established
-
Aged 12 years and older - Administer as in adults
The drug is best given twice daily. LEV is available in tablets of 250 mg, 500 mg, 750 mg, and 1000 mg. Intravenous (IV) and oral solutions are also available.
LEV is well tolerated. The most significant adverse effects are somnolence, asthenia, and dizziness. A number of patients reported infection, usually related to upper respiratory tract; however, none of these patients discontinued the drug, and it was not associated with changes in WBC count.
In the phase II studies and open extension studies that represent approximately 2258 patient-years exposure of to LEV, adverse effects were reported in more than 10% of cases. They included headache (25%), accidental injury (25%), convulsion (23%), infection (23%), asthenia (22%), somnolence (22%), dizziness (18%), pain (15%), pharyngitis (11%), and a flu-like syndrome (10%).
No serious acute idiosyncratic reactions have been reported, and no evidence of visual field disturbance has been reported. LEV has no strong tendency to exacerbate seizures, unlike this paradoxical effect recorded in some patients treated with other AEDs.
LEV is very useful in patients with hepatic or renal insufficiency and patients on concomitant medications, because it has no drug interactions. Current data support a good safety profile and efficacy in different patient populations, causing LEV to become one of the preferred AEDs in elderly patients. An IV preparation is available and a good alternative to treat seizures in the acute setting.
Efficacy in status epilepticus has not been established. The antiepileptogenic effect observed in kindling models makes it a potential agent for the prevention of epilepsy in conditions such as traumatic brain injury. However, no clinical studies have been performed to confirm this hypothesis.
Brivaracetam
The precise mechanism of action for brivaracetam (Briviact) is unknown. Brivaracetam displays a high and selective affinity for synaptic vesicle protein 2A (SV2A) in the brain, which may contribute to the anticonvulsant effect. It is indicated as monotherapy or adjunctive therapy for partial-onset seizures in adults and children aged 16 y or older.
Approval was based on results from a randomized, double-blind, placebo-controlled, multicenter study (n=768) that enrolled patients with uncontrolled POS despite ongoing treatment with 1-2 antiepileptic drugs. Percent reduction in POS compared with placebo was 22.8% and 23.2% for brivaracetam 100 mg/day and 200 mg/day respectively (p < 0.001). [93]
In September 2017, FDA approved brivaracetam (Brivact) as monotherapy for partial-onset (focal) seizures in patients aged ≥16 years with epilepsy. The supplemental New Drug Application (sNDA) approval was based on the FDA guidance that allows for extrapolation of the safety and efficacy of drugs approved as adjunctive therapy for treatment of partial-onset seizures. The sNDA submitted to support the monotherapy indication included clinical data involving over 2,400 adults with partial-onset seizures.
The starting dose is 50 mg PO BID. Based on individual patient tolerability and therapeutic response, adjust dose down to 25 mg BID (50 mg/day) or up to 100 mg BID (200 mg/day). Patient with any degree of hepatic impairment should be initiated at a lower dose (25 mg BID) and should not exceed a daily dose of 75 mg BID (150 mg/day).
Rufinamide
Rufinamide is a triazole derivative that is structurally unrelated to other AEDs approved in the United States. The precise mechanism of the antiepileptic effect is unknown. In vitro studies suggest that the principal mechanism of action of rufinamide is modulation of the activity of sodium channels and, in particular, prolongation of the inactive state of the channel.
It is indicated as an adjunctive treatment for seizures associated with Lennox-Gastaut syndrome in adults and children aged 1 y or older.
The initial adult dose is 400-800 mg/day PO divided q12h. Increase the daily dose by 400-800 mg every other day until a maximum of 3200 mg/day divided q12h is reached.
The initial pediatric dose is 10 mg/kg/d PO divided q12h. The pediatric target dose is 45 mg/kg/day divided q12h. Increase the daily dose by ~10 mg/kg every other day (not to exceed 3200 mg/day) until the target dose is reached.
It is not known whether doses lower than the target doses are effective.
Cannabidiol
A purified formulation of cannabidiol (Epidiolex) was approved in June 2018 for seizures associated with Lennox-Gastaut syndrome (LGS) or Dravet syndrome (DS). In 2020, it gained approval for seizures associated with tuberous sclerosis complex (TSC) and also gained approval for use in children as young as 1 year (originally approved for patients aged 2 years or older). Cannabidiol is a structurally novel anticonvulsant and the exact mechanism by which it produces anticonvulsant effects is unknown. It does not appear to exert its anticonvulsant effects through CB1 receptors, nor through voltage-gated sodium channels.
Approval for LGS and DS was based on results from several studies that compared adding cannabidiol added to conventional AEDs to placebo and the incidence of drop seizures from baseline. An international study of 225 patients with LGS (mean patient age 15 years) were randomized to receive cannabidiol 20 mg/kg/day or 10 mg/kg/day, or placebo over 14 weeks. During the 4-week baseline period, the median number of drop seizures was 85 in all groups combined. The median reduction from baseline in drop-seizure frequency per 28 days during the treatment period was 41.9% in the 20-mg cannabidiol group, 37.2% in the 10-mg group, and 17.2% in the placebo group. During treatment, 30 patients (39%) in the 20-mg group, 26 (36%) in the 10-mg group, and 11 (14%) in the placebo group had at least a 50% reduction from baseline in drop-seizure frequency. The odds ratio (OR) for 20 mg vs placebo was 3.85 (95% CI, 1.75 - 8.47; P < 0.001) and the OR for 10 mg vs placebo was 3.27 (95% CI, 1.47 - 7.26; P = 0.003). [94]
A study (n = 171) conducted in 24 clinical sites in the United States, the Netherlands, and Poland showed a median percentage reduction in monthly drop seizure frequency from baseline was 43.9% (IQR -69.6 to -1.9) in the cannabidiol group and 21.8% (IQR -45.7 to 1.7) in the placebo group. The estimated median difference between the treatment groups was -17.21 (P = 0.0135) during the 14-week treatment period. [95]
A double-blind, placebo controlled study found that investigated cannabidiol for the treatment of drug-resistant seizures in the Dravet syndrome found that the median frequency of convulsive seizures per month decreased from 12.4 to 5.9 win the cannabidiol group compared to a decrease from 14.9 to 14.1 in the placebo group. The adverse events that were more frequent in the cannabidiol group included diarrhea, vomiting, fatigue, pyrexia, somnolence, and abnormal results on liver-function test. [96]
Approval for use in TSC was based on the GWPCARE6 phase 3 trial (n = 224). Analysis of the 201 patients who completed the study showed that cannabidiol 25 mg/kg/day produced a significantly greater reduction in seizure frequency compared with placebo (49% vs 27%; P = .0009). [97]
Systemic exposure and adverse effects of cannabinol are dose-related. An open-label safety study by Gaston et al on the interactions between common antiepileptic drugs and the pharmaceutical formulation of cannabidiol (CBD; Epidiolex) reported that with increasing doses of cannabidiol, increases in topiramate, rufinamide, and N-desmethylclobazam and decrease in clobazam serum levels were seen (all p < 0.01). The study also found that in adults, increases in serum levels of zonisamide (p = 0.02) and eslicarbazepine (p = 0.04) were seen. Also of note was higher levels of AST/ALT in those who are also taking valproate. All changes were within the accepted therapeutic range. [98, 99]
Stiripentol
The FDA approved stiripentol (Diacomit) in August 2018 for treatment of seizures associated with Dravet syndrome in patients aged 2 years or older who are taking clobazam. This indication was expanded in July 2022 to include children as young as 6 months. There are no clinical data to support the use of stiripentol as monotherapy in Dravet syndrome.
Stiripentol belongs to the group of aromatic allylic alcohols and is unrelated to other anticonvulsants. The precise anticonvulsant effect in humans is unknown. Possible mechanisms of action include direct effects mediated through the GABA-A receptor and indirect effects involving inhibition of cytochrome P450 activity with resulting increase in blood levels of clobazam and its active metabolite.
Approval was based on 2 multicenter placebo-controlled double-blind randomized studies, conducted according to similar protocols. To be enrolled in either study, patients were required to be aged 3 years to less than 18 years, to have Dravet syndrome (ILAE classification of epilepsy, 1989), and to be inadequately controlled on clobazam and valproate, with at least 4 generalized clonic or tonic-clonic seizures per month despite optimized therapy. In Study 1 (N=41), 71% of stiripentol-treated patients were responders vs 5% in the placebo group (P < 0.0001), while in Study 2 (N=23), 67% of stiripentol-treated patients were responders vs 9.1% in the placebo arm (P < 0.0094). Treatment with stiripentol was also superior to placebo for the reduction in mean frequency of generalized clonic or tonic-clonic seizures. [100]
Fenfluramine
Fenfluramine is indicated for treatment of seizures associated with Dravet syndrome in patients aged 2 years and older.
Fenfluramine and its metabolite, norfenfluramine, increase extracellular levels of serotonin through interaction with serotonin transporter proteins and exhibit agonist activity at serotonin 5HT-2 receptors. The precise mechanism of action for the treatment of seizures associated with Dravet syndrome is unknown.
Approval was based on data from two phase 3 randomized, double-blind, placebo-controlled trials. When added to existing therapy, fenfluramine significantly reduced the monthly convulsive seizure frequency compared to placebo in study patients whose seizures were not adequately controlled on one or more antiepileptic drugs. Additionally, most study patients responded to treatment within 3-4 weeks, and effects remained consistent over the treatment period. [101, 102]
Neuronal Potassium Channel Openers
Ezogabine (Potiga), known as retigabine internationally, has a novel mechanism of action as a potassium channel opener. Ezogabine was approved by the US Food and Drug Administration (FDA) in June 2011. The FDA approved ezogabine as adjunctive therapy in partial-onset seizures uncontrolled by current medications.
A multicenter, randomized, double-blind, placebo-controlled trial evaluated the safety and efficacy of this agent. The study design was that of a typical adjunctive AED trial with a very refractory population of patients with localization-related epilepsy. [103]
In terms of both efficacy measures and tolerability, ezogabine appears comparable to prior second-generation AEDs, most recently lacosamide. However, because of its unique mechanism of action, it also comes with a different set of adverse effects. For example, potassium channels are expressed in smooth muscles, including the bladder and heart.
Ezogabine may be an option for patients whose condition is not controlled on their current medications; however, it remains to be determined how this agent will work with other medications. Like prior newer AEDs, ezogabine will not change the landscape of epilepsy or the fact that approximately 30% of patients with seizures are medically intractable. Clinicians should be open to trying new medications, and they should act on the recently updated definition of refractory epilepsy formulated by the International League Against Epilepsy (ILAE).
Questions & Answers
Overview
What are antiepileptic drugs (AEDs)?
What are the mechanisms of action and pharmacokinetics of antiepileptic drugs (AEDs)?
What are the mechanisms of action and pharmacokinetics of sodium channel blockers?
What are the mechanisms of action and pharmacokinetics of calcium channel blockers?
What are the mechanisms of action and pharmacokinetics of GABA enhancers?
What are the mechanisms of action and pharmacokinetics of glutamate receptor blockers?
What are the mechanisms of action and pharmacokinetics of carbonic anhydrase inhibitors?
What are the mechanisms of action and pharmacokinetics of sex hormones?
What are the mechanisms of action and pharmacokinetics of SV2A-binding agents?
What is the role of carbamazepine (CBZ) in the treatment of epilepsy?
What is the role of fosphenytoin in the treatment of epilepsy?
What is the role of oxcarbazepine (OXC) in the treatment of epilepsy?
What is the role of eslicarbazepine acetate (Aptiom) in the treatment of epilepsy?
What is the role of lamotrigine (LTG) in the treatment of epilepsy?
What is the role of zonisamide (ZNS) in the treatment of epilepsy?
What is the role of lacosamide in the treatment of epilepsy?
What is the role of sodium channel blockers in the treatment of epilepsy?
What is the role of phenytoin (PHT) in the treatment of epilepsy?
What is the role of clobazam in the treatment of epilepsy?
What is the role of clonazepam in the treatment of epilepsy?
What is the role of midazolam in the treatment of epilepsy?
What is the role of phenobarbital in the treatment of epilepsy?
What is the role of primidone in the treatment of epilepsy?
What is the role of benzodiazepines and barbiturates in the treatment of epilepsy?
What is the role of tiagabine (TGB) in the treatment of epilepsy?
What is the role of GABA reuptake inhibitors in the treatment of epilepsy?
What is the role of GABA transaminase inhibitors in the treatment of epilepsy?
What is the role of vigabatrin (VGB) in the treatment of epilepsy?
What is the role of gabapentin (GBP) in the treatment of epilepsy?
What is the role of pregabalin in the treatment of epilepsy?
What is the mechanism of action of valproate (VPA) and gabapentin (GBP)?
What is the role of valproate (VPA) in the treatment of epilepsy?
What is the role of felbamate in the treatment of epilepsy?
What is the role of topiramate in the treatment of epilepsy?
What is the role of glutamate blockers in the treatment of epilepsy?
What is the role of perampanel (Fycompa) in the treatment of epilepsy?
What is the role of levetiracetam (LEV) in the treatment of epilepsy?
What is the role of rufinamide in the treatment of epilepsy?
What is the role of stiripentol (Diacomit) in the treatment of epilepsy?
What is the role of brivaracetam (Briviact) in the treatment of epilepsy?
What is the role of cannabidiol (Epidiolex) in the treatment of epilepsy?
What is the role of ezogabine (Potiga) in the treatment of epilepsy?
-
Pearls of antiepileptic drug use and management.
-
Dynamic target of seizure control in management of epilepsy is achieving balance between factors that influence excitatory postsynaptic potential (EPSP) and those that influence inhibitory postsynaptic potential (IPSP).
-
Antiepileptic drugs can be grouped according to their major mechanism of action. Some antiepileptic drugs work by acting on combination of channels or through some unknown mechanism of action.
-
Some antiepileptic drugs stabilize inactive configuration of sodium (Na+) channel, preventing high-frequency neuronal firing.
-
Low-voltage calcium (Ca2+) currents (T-type) are responsible for rhythmic thalamocortical spike and wave patterns of generalized absence seizures. Some antiepileptic drugs lock these channels, inhibiting underlying slow depolarizations necessary to generate spike-wave bursts.
-
Gamma-aminobutyric acid (GABA)-A receptor mediates chloride (Cl-) influx, leading to hyperpolarization of cell and inhibition. Antiepileptic drugs may act to enhance Cl- influx or decrease GABA metabolism.
-
Glutamate (main excitatory neurotransmitter in central nervous system) binds to multiple receptor sites that differ in activation and inactivation time courses, desensitization kinetics, conductance, and ion permeability. Three main glutamate receptor subtypes are N-methyl-D-aspartate (NMDA), metabotropic, and non-NMDA (alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid [AMPA] and kainate receptors). Antiepileptic drugs known to possess this mechanism of action are listed.
-
Schematic representation of N-methyl-D-aspartate (NMDA) receptor.
-
GABA drugs and their known sites of action.
Tables

What would you like to print?
- Overview
- Mechanism of Action
- Sodium Channel Blockers
- GABA Receptor Agonists
- GABA Reuptake Inhibitors
- GABA Transaminase Inhibitors
- AEDs with Potential GABA Mechanism of Action
- Glutamate Blockers
- AEDs with Other Mechanisms of Action
- Neuronal Potassium Channel Openers
- Questions & Answers
- Show All
- Media Gallery
- References





