Clues to Diagnosis
Several primary neurodegenerative disorders distinct from Parkinson's disease (PD) share parkinsonian features of bradykinesia, rigidity, tremor, and balance disturbances. These disorders have complex clinical presentations that reflect degeneration in various neuronal systems. However, because of the common parkinsonian features, the disorders have been collectively named Parkinson-plus syndromes.
Parkinson-plus syndromes respond poorly to the standard treatments for PD. An inadequate response to treatment in a patient with parkinsonian symptoms suggests the possibility of Parkinson-plus syndrome and warrants a search for the signs and symptoms of degeneration in other neuronal systems.
In addition to a lack of response to carbidopa/levodopa or dopamine agonists in the early stages of the disease, other clinical clues suggestive of Parkinson-plus syndromes include the following:
-
Early onset of dementia
-
Early onset of postural instability causing frequent falls
-
Early onset of psychosis unrelated to medication side effects
-
Ocular signs, such as impaired vertical gaze, square-wave jerks, nystagmus, blepharospasm, and apraxia of eyelid opening or closure
-
Pyramidal tract signs not explained by previous stroke or spinal cord lesions
-
Severe autonomic symptoms such as postural hypotension and urinary incontinence early in the course of the disease
-
Prominent motor apraxia
-
Alien-limb phenomenon
-
Marked symmetry of signs in the early stages of the disease
-
Truncal symptoms more prominent than appendicular symptoms
-
Absence of secondary etiologies to explain the atypical findings (eg, normal pressure hydrocephalus)
Modern immunocytochemical techniques and genetic findings suggest that Parkinson-plus syndromes can be broadly grouped into 2 types: synucleinopathies and tauopathies. Clinically, however, 5 separate Parkinson-plus syndromes have been identified, as follows:
-
Multiple system atrophy (MSA)
-
Parkinsonism-dementia-amyotrophic lateral sclerosis complex
For more information, see the Medscape Reference article Parkinson's Disease.
See the related images below regarding Parkinson-plus syndromes.
 Alpha-synuclein staining of the pons of an MSA case showing the positive glial inclusions (40x).
Alpha-synuclein staining of the pons of an MSA case showing the positive glial inclusions (40x).
 Neuronal loss in the substantia nigra with pigment-laden macrophages and neuromelanin pigment spilled into the neuropil background (pigment incontinence).
Neuronal loss in the substantia nigra with pigment-laden macrophages and neuromelanin pigment spilled into the neuropil background (pigment incontinence).
 Tau-positive neuronal inclusions in neurons of the substantia nigra (no alpha synuclein-positive inclusions, as are found in Parkinson disease).
Tau-positive neuronal inclusions in neurons of the substantia nigra (no alpha synuclein-positive inclusions, as are found in Parkinson disease).
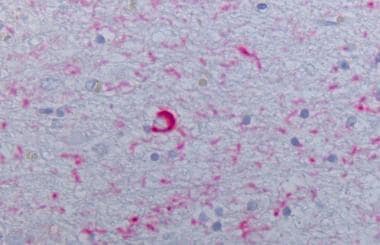
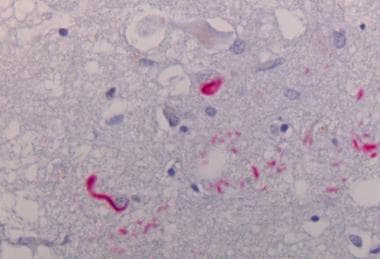
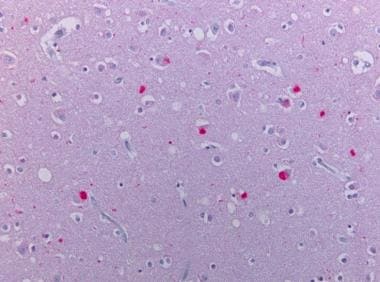 Neocortex stained alpha synuclein. The presence of cortical Lewy bodies is confirmed by the finding of alpha synuclein positive rounded cytoplasmic inclusion in neurons.
Neocortex stained alpha synuclein. The presence of cortical Lewy bodies is confirmed by the finding of alpha synuclein positive rounded cytoplasmic inclusion in neurons.
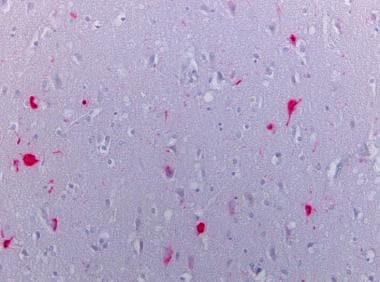
Multiple System Atrophy
The clinical definition of multiple system atrophy (MSA) is a progressive, idiopathic, degenerative process beginning in adulthood, manifesting in various degrees of autonomic failure, parkinsonism, cerebellar dysfunction, and pyramidal signs that are poorly responsive to levodopa or dopamine agonists. Glial cytoplasmic inclusions (GCIs) and neuronal multisystem degeneration are the pathologic hallmarks of this clinically variable disorder (see the image below).
Alpha-synuclein staining of the pons of an MSA case showing the positive glial inclusions (40x).
Dejerine and Thomas first used the term olivopontocerebellar atrophy (OPCA) in 1900 when they described 2 patients with a degenerative disorder leading to progressive cerebellar dysfunction and parkinsonism. In 1960, van de Eecken, Adams, and van Bogaert reported 3 patients with striatonigral degeneration (SND) with atrophy of the caudate nucleus and putamen. In 1960, Shy and Drager described a neurologic syndrome (Shy-Drager syndrome [SDS]) of orthostatic hypotension with parkinsonian features. SDS is identified by parkinsonism, which rarely responds to dopaminergic therapy and autonomic dysfunction. Though the main features of OPCA are cerebellar manifestations, mild parkinsonism and pyramidal signs are also typically present. Patients with SND usually display parkinsonism and pyramidal signs. Somewhat unique for SND is the laryngeal stridor. Not uncommonly, it is very difficult to distinguish SND from PD. [1] In 1969, Graham and Oppenheimer noted that the clinical and pathologic findings of OPCA, SND, and SDS overlapped significantly. They advanced the term multiple system atrophy to describe these disorders.
OPCA, SND, and SDS were considered distinct entities reflecting the degeneration of separate neuronal subsystems. Because patients shared many signs and symptoms, the clinical distinction may be unclear. Recent neurobiological research has justified the grouping of these conditions under a common pathophysiologic definition as variants of MSA.
Diagnosis
In the last 30 years a set of consensus diagnostic criteria for MSA have been developed. [2, 3, 4] These criteria suggest levels of certainty for MSA diagnosis: possible, probable, and definite. The definite diagnosis requires neuropathological confirmation. MSA is subdivided into 2 clinicopathological categories based on the neural system predominantly involved. Patients with predominant parkinsonian features are identified as having MSA-P, which replaces the term SND, whereas patients with prominent cerebellar dysfunction have MSA-C, which replaces the term OPCA. In a large portion of cases, however, both phenotypes and related pathologies are equally present. [5] Autonomic dysfunction appears in all forms of MSA, therefore, the term SDS is not useful. [3, 6]
MSA is most likely to be confused with idiopathic PD. Patients with idiopathic PD are distinguished from patients with MSA by the lack of severe autonomic and cerebellar features, as well as by their robust response to dopamine replacement therapy. In contrast, parkinsonian symptoms in MSA either do not respond to or require much higher doses of levodopa at the onset of therapy, and the efficacy decreases rapidly. In a prospective clinicopathologic study, the initial diagnosis of PD was correct in 65% of patients, a rate that improved to 76% with follow-up care. MSA was correctly diagnosed in 69% of patients who had been observed for at least 5 years. Severe autonomic insufficiency and cerebellar signs are the features most helpful for differential diagnosis. Some spinocerebellar ataxias (SCA) can manifest with clinical findings suggestive of MSA. SCA-3 and SCA-17 in particular can have these findings. [6]
Epidemiology
The exact incidence of MSA is not known; many experts believe that the disorder is underrecognized. Some authors estimate that 3–10% of patients diagnosed with PD actually have MSA-P, and a prevalence of 16.4 cases per 100,000 population has been reported. In a study in Minnesota from 1976 to 1990, researchers estimated the average annual incidence of MSA was 3 cases per 100,000 population. A study in rural Bavaria showed a prevalence of 0.31% in the population older than 65 years, a group in which 0.71% had PD.
MSA has a male predominance, as documented in 3 of 4 large studies. One study of 203 patients with histopathologically confirmed MSA demonstrated a male-to-female ratio of 1.3:1.0. The mean patient age at onset is 54.3 years, with a range of 33–78 years. [7]
Clinical presentation
There are four clinical domains in MSA:
-
Autonomic dysfunction
-
Parkinsonism
-
Cerebellar dysfunction
-
Corticospinal dysfunction
In a study of 100 patients with autopsy-proven MSA, initial symptoms were parkinsonism (46%), autonomic symptoms (41%), and cerebellar signs and symptoms (5%). [8] Nearly all patients eventually develop parkinsonism and autonomic symptoms. Orthostatic hypotension developed in 68% of patients. Cerebellar symptoms developed in 52% of patients during the course of the disease.
Autonomic failure manifests as urinary dysfunction progressing to complete incontinence, orthostatic hypotension, erectile dysfunction, or impotence, with the latter observed early in nearly all men with MSA. Common urinary manifestations are urinary frequency, urgency, incontinence, or incomplete bladder emptying. On electromyography (EMG), abnormal sphincteric results have been noted in MSA and can be useful ancillary findings. The early display of autonomic dysfunction is believed to anticipate a worse prognosis. [9, 10]
Patients with parkinsonism typically have asymmetric tremors, bradykinesia, rigidity, and severe and early postural instability. The tremor tends to be postural, irregular, and jerky, unlike the typical pill-rolling tremor of idiopathic parkinsonism. Prominent orofacial dyskinesia and dystonias are common upon treatment with levodopa. Antecollis dystonia is common in MSA. The dysarthria observed in patients with MSA tends to be hypokinetic. Those with cerebellar features present with gait and limb ataxia, ataxic dysarthria, square-wave jerks, and sustained gaze-evoked nystagmus. They also tend to develop saccadic pursuit movements.
Extensor plantar responses and hyperreflexia are present in patients with corticospinal dysfunction. Respiratory stridor is observed in 33% of patients; which may require a tracheostomy. Cognitive dysfunction is less common than in other Parkinson-plus syndromes, such as PSP or corticobasal degeneration (CBD).
Neuroimaging
Neuroimaging findings in patients with MSA correlate partially with the neuronal subsystems involved. All clinical subtypes tend to cause atrophy of the cerebellum, brainstem, putamen, and caudate nucleus. The globus pallidus tends to be spared in MSA. The cerebellum and brainstem tend to be atrophied in patients who present with predominantly cerebellar features, whereas the putamen and caudate nucleus tend to be involved in patients who present with parkinsonian features. Slitlike hyperintensities are noted on T2- and proton density–weighted MRIs of the pons, middle cerebellar peduncle, and cerebellum. An autopsy-proven case of MSA had hyperintensities in the pyramidal system on T2-weighted fluid-attenuated inversion recovery (FLAIR) MRI.
Correlations between MRI and histopathologic findings support the theory that iron deposition, microgliosis, astrocytosis, and severe neuronal loss may contribute to abnormal hyperintensities. The most reliable findings specific to MSA are putaminal atrophy, hyperintensity in the rim of the putamen, and infratentorial changes. However, these findings are not observed in all patients with MSA. The altered size of the inferior olivary nuclei, and putaminal isointensity or hypointensity relative to the globus pallidus are not useful findings. [11, 12, 13]
While the sensitive detection of putaminal iron deposition by T2*-weighted imaging (T2*-WI) is of diagnostic value for MSA, the diagnostic significance of the pontine hot-cross bun (HCB) sign remained unknown. Deguchi et al found that T2*-WI is of value for detecting the HCB sign, which might improve the diagnosis of MSA. [14]
Reduced metabolic activity in the putamen and decreased dopaminergic function in the striatonigral system have been demonstrated on positron emission tomography (PET) in patients with the parkinsonian subtype of MSA. However, these findings also are observed in idiopathic PD. Decreased metabolic activity in the cerebellum has been noted in the cerebellar subtype of MSA. PET studies may be useful in the differential diagnosis of MSA, PSP, and CBD. [15]
Routine MRI can be somewhat helpful in distinguishing MSA, PSP, and CBD. Putaminal involvement and vermian cerebellar atrophy are significantly more common in MSA, but cortical atrophy, midbrain atrophy, and third ventricle enlargement are more common in PSP and CBGD. The role of transcranial sonography in the investigation of these disorders is described below.
Pathophysiology
Although the exact cause of MSA evades understanding, many pathophysiologic mechanisms have been uncovered.
Iron and ferritin levels appear to be increased in the substantia nigra and striatum. Oligodendroglial and microglial cells are predominantly involved, with neurons and astrocytes relatively spared. Iron levels in the putamen are 5 times higher than normal and are associated with coarse electron-dense granules and fine granular and fibrillary material in lamellated structures. This excessive iron accumulation correlates with the signal voids noted in these structures on MRI. Excessive iron may produce neurotoxicity because of its role in oxidation and reduction reactions. This type of oxidative stress is postulated to be involved in various neurodegenerative disorders. Iron also promotes fibril formation from alpha-synuclein, which may be responsible for the formation of GCIs (see glial cytoplasmic inclusions, below).
Genetic susceptibility involving various genetic markers has been examined without confirmation. Genetic markers for previously identified spinocerebellar disorders are not found in patients with MSA. Mitochondrial chain function in the substantia nigra and platelets of patients with MSA are similar to those of age-matched controls.
Pathologic findings
MSA syndromes have common pathologic findings such as cell loss and gliosis in the striatum, substantia nigra, locus coeruleus, inferior olive, pontine nuclei, dorsal vagal nuclei, cerebellar Purkinje cells, and intermedio-lateral cell columns of the spinal cord. The specific histologic hallmark is glial cytoplasmic inclusions (GCI), which are found mainly in oligodendrocytes. [16]
Macroscopic findings in patients with MSA correlate with neuroimaging and clinical findings. Each MSA subtype demonstrates various degrees of atrophy in the extrapyramidal, spinocerebellar, pyramidal, and autonomic nervous systems. Some depigmentation of the substantia nigra and locus coeruleus is noted in all MSA subtypes. Atrophy of the motor and premotor cortices has also been noted. The intermediolateral cell column of the spinal cord is preferentially involved.
Patients with MSA-P primarily develop extrapyramidal system atrophy such that the posterolateral putamen and ventrolateral substantia nigra appear atrophic and discolored. Patients with MSA-C primarily have shrinkage of the cerebellum, middle cerebellar peduncles, inferior olives, and basis pontis. [17]
Histopathologic findings include neuronal loss, gliosis, and microvacuolation in the involved neuronal systems. These findings are present in varying degrees proportional to the volume loss on gross inspection. Even in regions where atrophy is not noted, these changes are present to some extent.
Demyelination eventually ensues in the white matter in the involved regions. An epitope of myelin basic protein was discovered only in degenerating myelin. Monoclonal antibody probes for this aberrant myelin demonstrate that these white matter lesions are more widespread than previously demonstrated with routine myelin stains.
Autonomic and endocrine manifestations of MSA may be related to neuronal loss in the hypothalamus, spinal cord, and medulla. Cell loss has been found in histaminergic neurons in the tuberomammillary nucleus, arginine-vasopressin neurons in the suprachiasmatic nucleus, and tyrosine hydroxylase (catecholamine-producing) neurons in the medulla, arcuate nucleus, and spinal cord lateral horns and intermediate zone of the anterior horns. Abnormalities in peripheral nerves and muscles that are absent in patients with idiopathic PD have been found in patients with MSA. Sural nerve biopsy demonstrates a 23% reduction in unmyelinated nerve fibers. Nerve conduction studies are abnormal in 40% of patients with MSA. Abnormal electromyography findings suggesting partial denervation have been found in 22.5% of patients with MSA. [18]
Neurodegeneration affecting axons may be a distinctive characteristic that can differentiate patients with MSA or PSP patients from patients with idiopathic PD. Neurofilament proteins reflecting axonal degeneration are increased in the CSF of patients with MSA or PSP.
Glial cytoplasmic inclusions
Microscopic findings in patients with MSA are distinctive for the cytoplasmic inclusions containing alpha-synuclein aggregates in the oligodendroglial cells, as well as for neuronal loss, astrocytosis, and loss of myelin. These lesions are predominantly located in the substantia nigra, locus coeruleus, putamen, inferior olives, pontine nuclei, Purkinje cells, and intermediolateral columns of the spinal cord. The globus pallidus, caudate nucleus, corticospinal tracts, anterior horn cells of the cord, dentate nucleus, and vestibular nuclei are relatively spared.
Many neurodegenerative disorders have been associated with a distinctive pathognomonic histopathologic lesion that can assist in diagnosis. Until 1989, no such distinctive lesion was associated with MSA, and the histopathologic diagnosis was based on nonspecific and variable neuronal-system atrophy, cell loss, myelin pallor, and astrocytosis. In 1989, GCIs were described in patients with MSA.
GCIs are argyrophilic and have various shapes (eg, triangular, sickle, half-moon, oval, conical). They are occasionally flame-shaped and may superficially resemble neurofibrillary tangles. However, cytoplasmic location, size, ultrastructure, immunocytochemical profile, and regional distribution of GCIs are distinctive. GCIs vary in size; they may fill the cytoplasm completely and push the nucleus to the side. The distribution of GCIs follows the suprasegmental motor system, supraspinal autonomic system, and their targets. This distribution includes the primary and secondary motor cortices, the pyramidal and extrapyramidal tracts, and the corticocerebellar systems.
The density of GCIs correlates with the severity of symptoms of patients with MSA. The distribution of GCIs correlates with the subtypes of MSA: putaminal lesions are prevalent in patients with the MSA-P subtype, and corticopontine lesions are prevalent in patients with the MSA-C subtype. Pyramidal lesions correlate with the severity of symptoms in both subtypes.
Ultrastructural studies of GCIs with electron microscopy and monoclonal antibody probes have confirmed their location in oligodendroglial cells and revealed them to be composed of ubiquitin, tau, microtubule-associated protein-5, cyclin-dependent kinase 5 (cdk5), mitogen-activated protein kinase (MAPK), and alpha-synuclein. The tau component in GCIs appears to be immunologically distinct from the tau protein found in patients with Alzheimer's disease, PSP, or corticobasal degeneration (CBD). MAPK and cdk5 are usually found in neurons and not oligodendroglial cells. Phosphorylation of these microtubular proteins of the cytoskeleton by these aberrantly located or expressed protein kinases may lead to the formation of GCIs.
Other neuronal inclusions have been observed in both the cytoplasm and nucleus of both oligodendroglial cells and neurons in patients with MSA. However, these findings are seen less commonly than GCIs, which remain the hallmark lesion of MSA. The density of GCIs appears to correlate with the severity of oligodendroglial degeneration and not with the potential degeneration of axons or neurons.
The finding of GCIs within oligodendroglial cells of patients with MSA has led to a shift in research interest from neurons to glial cells in patients with various neurodegenerative disorders. A variety of cellular alterations in the glial cells of patients with various neurodegenerative disorders has been described. However, none have resembled GCIs, and the clinical significance of these markers remains unclear. Further research in glial pathology may help uncover the pathophysiology in patients with neurodegenerative disorders.
Treatment and prognosis
Patients who develop MSA are routinely faced with a progressive disorder that eventually culminates in disability and death. Median survival in a study of 100 patients was 6.2 years, with a range of 0.5–24 years. Patients with the cerebellar subtype of MSA survived longer. Response to drug therapy is poor. The ataxia is particularly resistant to therapy. Dopamine replacement with levodopa is the mainstay of therapy for parkinsonian features (see carbidopa/levodopa). Responses tend to be less clear-cut, require higher doses, and may be transient in comparison to PD. Dopamine agonists are not effective and are more likely to cause hallucinations and psychosis. Blepharospasm and limb dystonia can be reduced with botulinum toxin injections.
Autonomic dysfunction, especially orthostatic hypotension, is a prominent feature of the disease. Initial treatment includes reduction of antihypertensive agents, increased salt and water intake, abdominal binders, and use of elastic stockings. Fludrocortisone and midodrine can be helpful when other measures fail. Other medications for the treatment of orthostatic hypotension include droxidopa and pyridostigmine. [19]
Progressive Supranuclear Palsy
Progressive supranuclear palsy (PSP) is reportedly the second most common cause of idiopathic parkinsonism after Parkinson's disease (PD). Generally, there is no family history or strong genetic component in this idiopathic condition. However, some rare familial clusters have been reported. The disease usually begins when patients are in their 50s to mid-60s. However, the youngest autopsy-proven case began when the patient was 43 years old. Approximately one-third of cases present when the patient is younger than 60 years.
In the Olmsted County Database, the average annual incidence for individuals aged 50–99 was 5.3 per 100,000 population. Overall, estimates suggest that PSP is about as common as motor neuron disease. A high incidence of PSP (and other atypical parkinsonism) has been reported in the French Antilles, approaching 14 cases per 100,000 population in Guadeloupe. This high incidence might be related to the local consumption of tropical fruit containing the mitochondrial toxin annonacin.
PSP is associated with neuronal loss, gliosis, and neurofibrillary tangles in the pretectal area, substantia nigra, subthalamic nucleus, globus pallidus, superior colliculus, and substantia innominata. Degeneration of multiple neurotransmitter systems leads to a more diffuse disorder than idiopathic PD. The cholinergic and adrenergic systems are involved in addition to the dopaminergic system. Tau-positive inclusions (tufted astrocytes and globose neurofibrillary tangles) in gray matter are consistent pathologic findings (see the images below). Coiled bodies, which are small round accumulations of tau in oligodendrocytes in the white matter, are also seen in a widespread distribution. [20, 21, 22, 23]
Abnormality of tau protein
PSP is considered to be a microtubule-associated protein tau (MAPT) disorder. [24]
Cortical fibrillary tangles of PSP are a pathological finding similar to that observed in Alzheimer's disease with regard to the presence of an abnormally phosphorylated tau protein. PSP also overlaps with corticobasal degeneration (CBD) in being a tauopathy. The mechanism whereby tau-protein pathology in PSP develops is yet to be determined.
Tau proteins exist in 6 isoforms encoded by a single gene. Due to alternate gene splicing of microtubule-binding site domains, protein isoforms containing 3 repeats (3R) or 4 repeats (4R) of these domains exist. In Alzheimer's disease, the pattern of tau deposits consists of a mix of 3R or 4R isoforms forming paired helical filament, whereas in PSP and CBD purely 4R isoforms are observed in straight filament conformation. Unlike in Alzheimer’s disease, no beta-amyloid deposits are seen in PSP. Tau protein abnormalities have also been found in frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). The chromosomal region containing MAPT has been shown to evolve into 2 major haplotypes: H1 and H2. The more common haplotype, H1, is overrepresented in patients with PSP and CBD. Genome-wide association studies found novel gene loci and common variants of MAPT associated with PSP. [25]
Thirty-two mutations of the tau protein have been identified in more than 100 families. [26] About half of the known mutations have their primary effect at the protein level. They reduce the ability of tau protein to interact with microtubules and increase its propensity to assemble into abnormal filaments. The other mutations have their primary effect at the RNA level and perturb the normal ratio of 3-repeat to 4-repeat tau isoforms. When studied, this change resulted in a relative overproduction of tau protein with 4 microtubule-binding domains in the brain. Familial PSP is extremely rare however, and the majority of cases are sporadic.
Clinical presentation and diagnosis
The onset of PSP typically begins in the sixth or seventh decade of life. The patient develops rapidly progressing postural instability leading to frequent falls, bradykinesia, rigidity, dysarthria, dysphagia, and, concurrently, frontal lobe dementia. Tremor is rare, and axial rigidity appears to be more prominent than limb rigidity. Consistently, patients have downgaze ophthalmoparesis. Eyelid dystonia is often present causing difficulty with either opening or closing the eyes. Dystonia of all types is present in about 13% of patients with pathologically proven PSP. [27]
The supranuclear component of the disorder is ocular paresis, which can be overcome by the vertical doll's-eyes maneuver. The combination of vertical gaze paresis and a history of frequent falls due to postural instability is central to the diagnosis of PSP. Bilateral frontal lobe dysfunction frequently manifests in palilalia, emotional incontinence (pseudobulbar affect), presence of applause sign on clinical examination, [28] and other signs of frontal lobe degeneration. Neuropsychiatric features include apathy and disinhibition; visual hallucinations are not common.
The National Institute of Neurological Disorders and Stroke and the Foundation for PSP/CBD and Related Brain Diseases (CurePSP) have published formal criteria for the diagnosis of PSP-Richardson syndrome. [25] Having both a vertical supranuclear palsy and postural instability is indicative of clinically probable PSP, whereas having either an isolated vertical gaze palsy or slowed saccades with postural instability is indicative of clinically possible PSP, according to these criteria. Movement disorders society PSP consensus criteria, which are more sensitive but less specific and reflective of various phenotypes of PSP, were more recently generated by the MDS-PSP study group, based on the analysis of a large body of clinical literature and autopsy-confirmed cases. [29] As with other atypical parkinsonian disorders (MSA and CBGD) there are levels of diagnostic confidence suggested by both of these criteria sets including possible, probable, and definite PSP, with the latter category requiring neuropathological confirmation. Core clinical features of the disease according to the MDS criteria fall into four functional domains: oculomotor dysfunction, postural instability, akinesia, and cognitive dysfunction. Each domain’s present clinical features are scored according to three levels of diagnostic certainty (high, medium, low) and a combination of the domain scores contributes to overall diagnostic certainty (possible, probable). Postural instability leading to falls in the first year of disease onset and a vertical supranuclear gaze paresis have good discriminatory value for diagnosis. The most characteristic feature of PSP is supranuclear downgaze palsy. However, some patients who never develop this finding are found to have PSP at autopsy. There is an average delay in making a proper diagnosis of 3.6 years after symptom onset.
The two most common clinical subtypes of PSP are described. [30, 4] The first, called Richardson syndrome, is associated with an early onset of postural instability, supranuclear gaze palsy, and cognitive dysfunction. The second, called PSP-Parkinsonism, has a more protracted course, with features initially more typical of idiopathic PD, including a moderate response to levodopa, and oculomotor features developing later in the disease course. More rarely, patients present with freezing of gait, akinesia of speech, and handwriting, but without rigidity, tremor, dementia, or gaze paresis (PSP-progressive gait freezing variant). There are also other rare clinical forms of the disease, with presentations intersecting with other neurodegenerative syndromes, such as PSP with predominant speech and language disorder, PSP with predominant frontotemporal dementia, and PSP with predominant cortico-basal syndrome; or with presentation limited to only one typical core domain, such as PSP with predominant oculomotor dysfunction, PSP with predominant postural instability, and others.
MRI of the brain may be helpful in diagnosis, but may also be normal. Atrophy in the midbrain or cerebellum, whether general or focal, is the most common. Due to disproportionate atrophy of the dorsal midbrain relative to the pons, sagittal views on MRI can give the appearance of a "hummingbird." Still, up to a quarter of patients with PSP have no abnormality on computed tomography (CT) or MRI imaging of the brain. [31] Midbrain diameter of less than 17 mm, hyperintensity in the midbrain, atrophy or hyperintensity of the red nucleus, and hyperintensity in the globus pallidus are useful findings. The measurement of the midbrain atrophy ratio on MRI is a useful tool in distinguishing early PSP from PD and age-matched controls. [32, 33]
PET and single-photon emission CT (SPECT) demonstrate prefrontal hypometabolism. Specialized PET and SPECT scans can depict severe involvement of the dopaminergic system.
Treatment
Pharmacologic agents remain the mainstay of treatment of PSP, though the results are frequently disappointing.
Carbidopa/levodopa reduces bradykinesia and rigidity in perhaps one-third of patients. However, even at high doses dopaminergic therapies usually provide only a mild, temporary improvement of parkinsonian symptoms. This is likely due to the loss of dopamine receptors in the striatum. At present, no medication provides continued relief in such patients. [34] Dopamine agonists rarely help and are most likely to cause hallucinations and confusion. Sometimes, the reduction of bradykinesia may lead to an increase in falling because of postural instability. Therefore, physical therapy and rehabilitation should focus on gait training, and the use of assistive devices such as walkers should be considered. [35]
Emotional incontinence may respond to dextromethorphan/quinidine. A small controlled study of donepezil reported modest benefits for the cognitive symptoms at the expense of worsening activities of daily living (ADL) and motor scores. [36] Zolpidem has been reported to improve both motor symptoms and ocular findings, but this effect was transient. [37] Patients should be assessed for depression, and antidepressant medications can be used. Insomnia is common and can be treated with melatonin, trazodone, mirtazapine, and clonazepam. Severe agitation may require the use of antipsychotic medications, which, however, carry an increased overall mortality risk.
Blepharospasm can be treated with botulinum toxin injections. Botulinum toxin injections can also be useful if drooling of saliva is a disabling symptom. Dry eyes can be treated with topical lubricants. Monitoring for swallowing difficulties with a modified barium swallow test allows for appropriate dietary modification or percutaneous endoscopic gastrostomy tube placement to reduce aspiration risk.
Prognosis
Patients with PSP tend to have progressive deterioration, with a 9.7-year median survival from the onset of symptoms. Gait difficulties occur early, and patients require assistance within 3 years. Confinement to bed or a wheelchair is typically necessary within 8 years. Eventual death usually follows a severe fall, pulmonary embolus, or aspiration pneumonia.
Corticobasal Degeneration
Corticobasal degeneration (CBD), also known as cortical-basal ganglionic degeneration, is the rarest of the three main Parkinson’s-plus syndromes (progressive supranuclear palsy (PSP) and multiple system atrophy (MSA) being the other two). It is characterized by frontoparietal cortical atrophy in addition to degeneration within the extrapyramidal system. CBD patients exhibit cortical findings such as pyramidal signs, aphasia, unilateral cortical sensory deficits, neglect, orobuccal and limb apraxia, limb myoclonus, and alien limb phenomena, along with subcortical findings of dystonia and parkinsonism. In recent studies, there is increasing evidence of an overlapping presentation in some patients of progressive aphasia, supranuclear gaze palsy (mimicking PSP), and frontotemporal dementia. The main neuropathological findings include swollen achromatic neurons, gliosis, and neuronal loss in the cerebral cortex, substantia nigra, lateral nuclei of the thalamus, locus coeruleus, striatum, and Purkinje layer of the cerebellum. The disease tends to occur in those aged 60–80 years, with a mean age of onset of about 64 years. The estimated incidence is 0.4 per 100,000 population per year. Rare familial cases with a mutation of MAPT have been reported, as well as sporadic cases with known MAPT mutation, but most cases are sporadic without known genetic cause.
PSP and MSA may initially be confused with CBD. However, the true diagnosis becomes clear as apraxia and dystonia develop. The clinical and pathologic features of PSP and CBD can overlap considerably. [38, 39, 40, 41]
Cortical atrophy of the frontal and parietal lobes has been described, with ballooned (see the image below) and enlarged cells seen on microscopy. The substantia nigra is depigmented. However, Lewy bodies and diffuse neuronal loss are conspicuously absent.
Pathology
CBD is now classified as a 4-repeat tauopathy. [42] Neuropathologic diagnostic criteria include 4R tau-immunoreactive lesions in neurons, glia, and cell processes. The minimal pathologic features for diagnosis are cortical and striatal tau-positive glial inclusions, specifically astrocytic plaques, as well as less specific neuronal tau inclusions, threads, and oligodendroglial coiled bodies in both white and gray matter combined with neuronal loss in both focal cortical lesions and substantia nigra. These criteria help distinguish the disease from other tauopathies. Of note, patients meeting these pathologic criteria may not always have the classic clinical presentation of CBD.
Clinical presentation
The clinical onset of CBD occurs in the sixth decade of life. Rinne et al reported 5 initial presentations, including a "useless" arm (55%), gait disorder (27%), prominent sensory symptoms including astereognosis, isolated speech disturbance, and behavioral and psychiatric disturbances. [43]
Symptoms on long-term follow-up include focal or asymmetric rigidity, marked limb dystonia (“claw hand”), bradykinesia including parkinsonian gait, jerky postural and action tremor, and/or myoclonus. These problems usually arise predominantly in one upper extremity, although, more rarely, initial presentation in the lower limbs also occurs. Limb apraxia may become a serious problem, with involuntary limb movements occasionally as severe as alien limb phenomena. The incidence of limb apraxia is far higher in CBD than in PSP, with a similar level of cognitive impairment.
The most recent consensus set of diagnostic criteria [44] distinguishes 5 clinical phenotypes: possible and probable cortico-basal syndrome (CBS), frontal-behavioral spatial syndrome, nonfluent agrammatic variant of primary progressive aphasia syndrome, and progressive supranuclear palsy-like syndrome. Exclusion criteria differentiate CBD from PD, MSA, DLB, and other neurodegenerative conditions.
Treatment and prognosis
The rigidity, bradykinesia, and tremor in a minority of patients may benefit from levodopa therapy. However, the marked disability from limb apraxia is progressive and generally remains unresponsive to rehabilitation efforts. Injections of botulinum toxin may partially relieve dystonia. In particular, the dystonic clenched fist may respond to injections of botulinum toxin, which helps relieve the pain and prevent skin breakdown. CBGD usually progresses to severe disability and death within 10 years (mean estimated survival is 6.5 years).
Dementia with Lewy Bodies (DLB)
Dementia with Lewy bodies (DLB) is a progressive neurodegenerative disorder characterized by dementia preceding or coinciding with the onset of parkinsonian symptoms, commonly with prominent neuropsychiatric disturbances. It is the second most common type of dementia after Alzheimer’s disease, comprising up to 1 in every 13 patients with dementia in secondary care. [45] Progressive dementia is often the first and predominant symptom. Of note, longitudinal studies show that after a decade of motor symptoms, 78% of patients with Parkinson's disease (PD) meet the criteria for dementia, [46] as defined in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, Text Revision (DSM-5-TR). DLB and Parkinson's disease dementia (PDD) together represent a clinical spectrum of cognitive dysfunction caused by Lewy body pathology (under the combined term of Lewy body dementia). [47]
See the images below showing cortical Lewy bodies.
Neocortex stained alpha synuclein. The presence of cortical Lewy bodies is confirmed by the finding of alpha synuclein positive rounded cytoplasmic inclusion in neurons.
No familial disposition for DLB has been reported. The common thread between DLB and PD is the presence of Lewy bodies. These are rounded inclusion bodies containing alpha-synuclein as the main component, in association with other proteins, such as ubiquitin. In PD, they are mainly observed in the substantia nigra and only spread to the cortex in advanced disease, causing symptoms of PDD. In contrast, in DLB they are scattered throughout the cerebral cortex early in the disease and are also seen in the substantia nigra and other subcortical regions. In contrast to Alzheimer's disease and CBD, cortical atrophy is not prominent. Two distinct forms of DLB brain pathology have been described. The “pure” form occurs with Lewy bodies in the cortical and subcortical structures, whereas “the mixed pathology” form has both Lewy bodies and Alzheimer’s type neuritic amyloid-beta protein containing plaques and tau-protein containing neurofibrillary tangles. The combined AD and Lewy body pathology has been increasingly recognized.
Clinical features
The Consortium on Dementia with Lewy bodies consensus clinical and pathologic criteria for diagnosis of DLB (ie, DLB criteria), first proposed in 1996, are widely used. [48, 49, 50] The criteria allow for clinical diagnosis of DLB as possible and probable. The essential requirement for the diagnosis of all levels or certainty is the presence of dementia, which in DLB may include deficits of attention, executive function, and visual-perceptual ability early in the disease, while memory impairment is often a later feature. Core clinical features are (1) visual hallucinations, (2) fluctuating cognition, (3) REM-sleep behavior disorder that may precede cognitive decline, and (4) one or more spontaneous motor features of parkinsonism (resting tremor, bradykinesia, rigidity). Indicative biomarkers for DLB used by the criteria include evidence of dopamine deficiency in basal ganglia on SPECT or PET scan, absence of REM sleep atonia on polysomnogram, and reduced cardiac sympathetic innervation as evidenced by 123iodine-MIBG scintigraphy. One of the above four core features or the presence of at least one of the above indicative biomarkers is required to diagnose possible DLB. Two core features are needed for probable DLB diagnosis, or one core feature plus one indicative biomarker. The supportive clinical features include repeated falls, postural instability, syncope, other transient loss of consciousness, severe dysautonomia, severe neuroleptic sensitivity, systematized delusions, hallucinations in modalities other than visual (auditory, olfactory, tactile), hypersomnia, hyposmia, apathy, anxiety, and depression. The criteria also describe clinical features that make alternative diagnoses such as vascular dementia, Alzheimer’s dementia with parkinsonian motor features, or mixed pathology more likely. In DLB, dementia occurs before or concurrently with parkinsonism. A useful general clinical rule is that if dementia onset follows parkinsonism later than by 1 year, then the diagnosis of PDD rather than DLB should be used.
Approximately 15% of patients do not have any parkinsonian features. Neuropsychological deficits that have been described include aphasia, dyscalculia, and apraxia. Visual hallucinations are seen in 80% of cases. DLB patients are more likely to have cognitive adverse effects of levodopa therapy in the early stages than patients with PD.
Differential diagnosis
PDD and other dementias where parkinsonian signs may be present (eg, vascular dementia) are the entities to be considered. Presentation of Alzheimer's disease with parkinsonian signs may resemble DLB. Neuroimaging can be used in support of the diagnosis but is not definitive. Ioflupane-123I dopamine transporter SPECT scan helps to assess presynaptic dopamine deficiency and has been approved as an adjunct test for DLB by the US Food and Drug Administration. Occipital hypometabolism is a characteristic feature of DLB on PET scans, [51, 52] although this is not always present. [53] Preservation of metabolism in the middle and posterior cingulate cortex known as the “cingulate island” sign is a feature of DLB, distinguishing it from AD. [54] Medial temporal lobe structures are relatively preserved in DLB, while atrophy is detected in Alzheimer's disease, as found on brain CT/MRI. [55] Specific EEG characteristics of DLB not present in Alzheimer's disease exist and are currently explored as diagnostic tools. [56]
Treatment
Parkinsonian features may respond somewhat to levodopa therapy in some patients. Worsening hallucinations, confusion, and orthostatic hypotension are factors severely limiting levodopa use in DLB. Emphasis should be on fall prevention, with the use of assistive devices when necessary.
Centrally acting cholinesterase inhibitors (rivastigmine, donepezil [57] for which the evidence is the strongest, galantamine) partially reverse decreased cortical cholinergic activity and may improve cognition and neuropsychiatric symptoms in DLB. Rivastigmine improves cognition and neuropsychiatric symptoms in patients with DLB without worsening parkinsonian features.
Autonomic symptoms can be prominent and should be addressed; this includes both pharmacologic and non-pharmacologic management of orthostatic hypotension, constipation, and urinary symptoms. Sialorrhea can be effectively treated with botulinum toxin injections to salivary glands or low-dose oral (glycopyrrolate) or sublingual (atropine) anticholinergic medications with caution not to exacerbate confusion.
REM-sleep behavior disorder may require treatment; measures to prevent injury during sleep should be taken and medications such as melatonin and clonazepam can be used.
Newer generation neuroleptic medications (eg, quetiapine and even more so clozapine) may be effective in controlling psychosis without exacerbating parkinsonian symptoms. However, elderly patients with dementia-related psychosis who are treated with antipsychotic drugs are at increased risk of death, as shown in short-term controlled trials. Deaths in these trials appeared to be either cardiovascular (eg, heart failure, sudden death), cerebrovascular (stroke), or infectious (eg, pneumonia) in nature. Patients or caregivers should be informed of these risks before starting therapy.
Pimavanserin (Nuplazid) was approved in April 2016 for the treatment of hallucinations and delusions associated with Parkinson's disease psychosis. It is the first drug to be approved for this condition. Pimavanserin is a selective serotonin inverse agonist (SSIA) that preferentially targets 5-HT2A receptors but avoids activity at dopamine and other receptors commonly targeted by antipsychotics. Pimavanserin is not approved for the treatment of patients with dementia-related psychosis unrelated to the hallucinations and delusions associated with Parkinson's disease psychosis. Efficacy was shown in a 6-week clinical trial (n = 199), in which pimavanserin proved to be superior to placebo in decreasing the frequency and/or severity of hallucinations and delusions without worsening the primary motor Parkinson's disease symptoms (p = 0.001). [58] However, there is insufficient data to recommend the use of pimavanserin in DLB at present.
Prognosis is poor, as with the other syndromes already described: median survival estimates are about 4 years post diagnosis, but both more rapid (survival less than 1 year post diagnosis) and more prolonged (survival more than 7 years post diagnosis) forms exist. Failure to thrive and dysphagia leading to aspiration pneumonia are the most common immediate causes of death. Depression is common, leading to suicide in up to 1%. Recognition and treatment of depression and anxiety are important parts of management.
Parkinsonism-Dementia-ALS Complex
The Parkinsonism–dementia–amyotrophic lateral sclerosis complex (PDALS) is a condition well described on the island of Guam and is known there as Lytico-Bodig disease. The latter term is derived from the local Guamanian dialect, with “Lytico” referring to the paralysis caused by the ALS component and with “Bodig" referring to the "laziness" that describes the parkinsonian component.
Extensive genetic and environmental research has been performed on this disorder in the last 50 years. The incidence of PDALS peaked in the 1950s and has declined since then. Dietary toxins in native flour were once considered the source of a potential neurotoxin. However, this hypothesis has been ruled out. Flour is obtained from the seeds of the cycad tree. Because the seed contains a potent hepatotoxin, the flour must be washed many times before consumption. Cycasin and beta-N -methyl-amino-L-alanine (BMAA) are putative neurotoxins in the seed. If the seeds are repeatedly washed, ingestion of an estimated 70 kg of flour is required to receive a toxic dose; therefore, this hypothesis is unlikely. The toxic effects of manganese and aluminum are also being considered.
Another term used in this setting is disinhibition–dementia–parkinsonism–amyotrophy complex. However, this term is not confined to Guam and may represent tauopathy of the FTDP-17 type.
Pathologic evaluation of the substantia nigra has found depigmentation, basophilic inclusion bodies, cell loss, and neurofibrillary tangles without senile plaques. This last finding also has been observed in the anterior horn cells and pyramidal tracts. Motor neuron disease tends to occur in younger patients, and older patients tend to develop parkinsonism and severe dementia. In Guam, the presentation varies between the 2 cultural groups that have populated the island. Both the ALS form and the parkinsonism-dementia form of PDALS are seen in the Chamorros group, while the Filipino group tends to have the parkinsonism-dementia syndrome.
Consider Alzheimer's disease, Lewy body disease, postencephalitic parkinsonism, idiopathic PD, ALS, and motor neuron disorders in the differential diagnosis of PDALS. Regarding treatment, patients do not respond to levodopa, and psychiatric treatment may be indicated. A progressive clinical picture is characteristic of both subtypes of PDALS. Death within 10 years is usual.
Diagnosis and Management of Parkinson-Plus Syndromes
Patients with parkinsonian syndromes unresponsive to dopamine agonists or levodopa should have an imaging study of the brain. MRI of the brain is preferred to CT, as it provides better visualization of midbrain and brainstem structures. A specialized brain SPECT scan for dopamine transporter using radiopharmaceutical Ioflupane-123I has become a helpful adjunct diagnostic tool to confirm the state of presynaptic dopamine deficiency present in Parkinson's disease (PD) and Parkinson-plus disorders; however, it cannot differentiate between these conditions.
PET imaging with fluorine-18-fluorodeoxyglucose (FDG) has been used to identify characteristic patterns of regional glucose metabolism in patients with idiopathic PD or variants of parkinsonism, such as multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD). In a longitudinal study, Eckert et al compared FDG-PET diagnosis with a clinical diagnosis by a movement disorders specialist with a 2-year follow-up. Diagnoses based on a blinded computerized assessment of PET scans agreed with clinical diagnosis in 92.4% of all subjects (97.7% with early PD, 91.6% with late PD, 96% with MSA, 85% with PSP, 90.1% with CBGD) and in 86.5% of healthy control subjects. [59] Transcranial sonography (TCS) has been used as a tool for the differential diagnosis of these syndromes. Characteristic findings include hyperechogenicity in the substantia nigra in PD, while hyperechogenicity in the lentiform nucleus is seen in MSA and PSP. [60, 61] Walter et al observed that lenticular nuclei hyperechogenicity is observed in 72–82% of patients with MSA and PSP but in only 10–25% of patients with PD.
Magnetic resonance diffusion-weighted imaging regional apparent diffusion coefficients (rADC) may also be helpful. Middle cerebellar peduncle and rostral pons rADC in MSA are significantly greater than in PSP and PD. Pavior et al reported that middle cerebellar peduncle rADC distinguishes MSA from PSP with a sensitivity of 91% and a specificity of 84%. [62]
Comprehensive face-to-face counseling with the patient and caregivers is indicated. Although the relatively poor prognosis must be outlined, it is also important to emphasize positive supportive measures. These include the use of appropriate walkers (preferably with wheels and brakes), occupational therapy, assessment of the home environment, physical and occupational therapy, and exercise. Ask specifically about swallowing ability and, when indicated, obtain a modified barium swallow study.
When difficulties with aspiration are detected, consider a change in diet and, in advanced cases, placement of a percutaneous endoscopic gastrostomy tube to maintain nutritional status. Patients may exhibit an interest in surgical treatments, such as deep brain stimulation, pallidotomy, or transplantation, but these are not indicated for Parkinson-plus syndromes.
Levodopa and dopamine agonists
In general, patients with Parkinson-plus syndromes do not tolerate dopamine agonists well. The mainstay is a trial of levodopa at doses higher than those commonly used in PD. A typical regimen begins with carbidopa/levodopa 25/100 twice daily, with the dose increased by 0.5–1 tablet every week to a target daily dose of approximately 1000 mg of levodopa.
If adverse effects occur at any point in the titration, lower the dose and reassess the plan. Although most patients do not have a positive response, some may respond to high doses, with reduced rigidity, easing of transfers, or mild improvements in balance or gait. If high doses of levodopa do not help the patient, gradually lower the dose with the aim to discontinue the medication. Some patients may find that a low dose helps them stay mobile or improves rigidity, and this dose can be maintained. The patient and caregiver should have easy access to the physician during the trial of levodopa.
Treatment of cognitive problems
Dementia is the most difficult symptom to treat in this setting. Donepezil, rivastigmine, and galantamine can be tried but do not have any dramatic effect and sometimes can worsen motor symptoms. A comprehensive neuropsychological evaluation repeated at intervals of 1 year can help in assessing the progression of cognitive symptoms in a quantitative way and can suggest coping methods.
Psychosis and severe agitation can be alleviated by neuroleptics preferably with the least basal ganglia D2 dopamine-receptor blocking properties such as quetiapine and clozapine, but other, especially older generation neuroleptics can cause a marked increase in parkinsonism and are best avoided. The use of neuroleptics in patients with dementia carries an elevated risk of death due to cardiovascular causes, and patients and families should be informed of this before initiating treatment.
Surgical treatment
An exciting development is a possible application of deep brain stimulation (DBS) to Parkinson-plus syndromes. In an open-label trial, Stefani et al reported an improvement in gait and balance in patients with PSP undergoing DBS with the pedunculopontine nucleus and the subthalamic nucleus in combination. [63] Further studies are warranted to validate this result.
Treatment of eye involvement
Some patients with PSP may benefit from prismatic lenses that compensate to some extent for vertical gaze problems. Blepharospasm, facial dystonia, and apraxia of eyelid opening may respond to botulinum toxin injections.
Questions & Answers
Overview
What are Parkinson-plus syndromes?
What are the signs and symptoms of Parkinson-plus syndromes?
How are Parkinson-plus syndromes characterized?
What is multiple system atrophy (MSA)?
How is multiple system atrophy (MSA) diagnosed?
What is the prevalence of multiple system atrophy (MSA)?
What are the clinical domains of multiple system atrophy (MSA)?
What are the signs and symptoms of multiple system atrophy (MSA)?
Which neuroimaging findings are characteristic of multiple system atrophy (MSA)?
What is the role of MRI in the diagnosis of multiple system atrophy (MSA)?
What is the pathophysiology of multiple system atrophy (MSA)?
Which pathologic findings indicate multiple system atrophy (MSA)?
Which microscopic findings are characteristic of multiple system atrophy (MSA)?
What is the pathognomonic histopathologic lesion of multiple system atrophy (MSA)?
What are glial cytoplasmic inclusions (GCIs) in patients with multiple system atrophy (MSA)?
What are the treatment options and prognosis of multiple system atrophy (MSA)?
What is progressive supranuclear palsy (PSP)?
What is the incidence of progressive supranuclear palsy (PSP)?
What is the pathophysiology of supranuclear palsy (PSP)?
What is the role of tau protein in the pathophysiology of progressive supranuclear palsy (PSP)?
What are the signs and symptoms of progressive supranuclear palsy (PSP)?
What are the criteria for diagnosis of progressive supranuclear palsy (PSP)?
What are the clinical subtypes of progressive supranuclear palsy (PSP)?
What is the role of imaging studies in the diagnosis of progressive supranuclear palsy (PSP)?
What are the treatment options for progressive supranuclear palsy (PSP)?
What is the prognosis of progressive supranuclear palsy (PSP)?
What is corticobasal degeneration (CBD)?
Which pathological findings indicate corticobasal degeneration (CBD)?
What are the signs and symptoms of corticobasal degeneration (CBD)?
What is the treatment and prognosis of corticobasal degeneration (CBD)?
What is dementia with Lewy bodies (DLB)?
What is the pathophysiology of dementia with Lewy bodies (DLB)?
What are the signs and symptoms of dementia with Lewy bodies (DLB)?
How is dementia with Lewy bodies (DLB) diagnosed?
What is the differential diagnosis for dementia with Lewy bodies (DLB)?
What are the treatment options for dementia with Lewy bodies (DLB)?
What is parkinsonism – dementia – amyotrophic lateral sclerosis complex (PDALS)?
What is the prognosis parkinsonism – dementia – amyotrophic lateral sclerosis complex (PDALS)?
What is included in supportive care for Parkinson-plus syndromes?
What is the role of levodopa and dopamine agonists in the management of Parkinson-plus syndromes?
How are hallucinations and delusions managed in patients with Parkinson-plus syndromes?
What is the role of prismatic lenses in the management of Parkinson-plus syndromes?
What is the role of imaging studies in the management of Parkinson-plus syndromes?
What is the role of deep brain stimulation (DBS) in the management of Parkinson-plus syndromes?
-
Alpha-synuclein staining of the pons of an MSA case showing the positive glial inclusions (40x).
-
Rounded tau positive globose tangles in neurons of the subthalamic nucleus.
-
Neuronal loss in the substantia nigra with pigment-laden macrophages and neuromelanin pigment spilled into the neuropil background (pigment incontinence).
-
Tau-positive neuronal inclusions in neurons of the substantia nigra (no alpha synuclein-positive inclusions, as are found in Parkinson disease).
-
Subcortical white matter showing tau-positive perinuclear glial inclusions.
-
Subcortical white matter showing tau-positive perinuclear glial inclusions.
-
Cerebral cortex with ballooned neuron.
-
Cerebral cortex with tau-reactive cellular inclusions and neuropil threads.
-
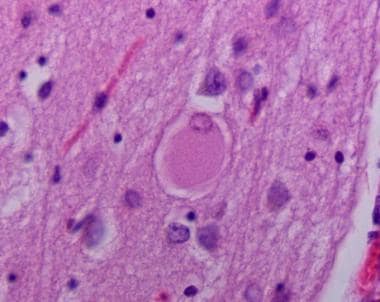
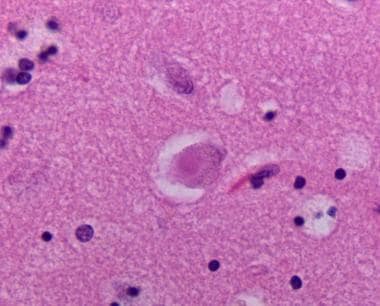
Hematoxylin and eosin stained section of neocortex showing cortical Lewy body.
-
Neocortex stained alpha synuclein. The presence of cortical Lewy bodies is confirmed by the finding of alpha synuclein positive rounded cytoplasmic inclusion in neurons.
-
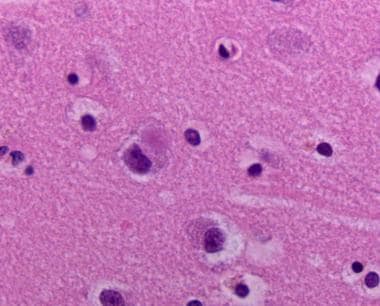
Neocortex stained with tau. Tau positive tangles in neurons of the cortex.








