Background
The association of anemia and gastrointestinal and neurologic abnormalities referable to the brain, spinal cord, and peripheral nerves has been recognized in several clinical and postmortem case reports and series by Combe, Addison, and Fenwick since the early 19th century. In 1877, Gardner and Osler coined the term pernicious anemia (PA) to describe a patient with progressive arm numbness and difficulty with buttoning and using tools. [1] Liechtenstein in 1884 reported the association of PA and spinal cord disease but attributed both to tabes dorsalis. [2] Lichtheim in 1887 [3] and Minnich in 1892 [4] recognized the histologic differences in the spinal cord between PA and tabes dorsalis.
In 1900, Russell et al coined the term subacute combined degeneration of the spinal cord. [5] In 1926, Minot and Murphy fed PA patients a half-pound of calf liver daily, for which they received the Nobel Prize. [6] In 1929, Castle distinguished the role of gastric (intrinsic) and dietary (extrinsic) factors in PA. [7] In 1948, cyanocobalamin was isolated from the liver. The existence of vitamin B-12 deficiency neuropathy was recognized in 1958. In 1955, Lassen et al [8] noted megaloblastic anemia secondary to prolonged nitrous oxide (N2 O) exposure; the neurologic features were described in 1978 by Sahenk et al [9] and Layzer et al. [10]
See the image below.
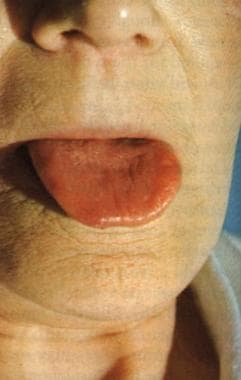 Vitamin B-12–associated neurological diseases. Pernicious anemia. Characteristic lemon-yellow pallor with raw beef tongue lacking filiform papillae. Photo from Forbes and Jackson with permission.
Vitamin B-12–associated neurological diseases. Pernicious anemia. Characteristic lemon-yellow pallor with raw beef tongue lacking filiform papillae. Photo from Forbes and Jackson with permission.
Pathophysiology
Vitamin B-12 structure
Vitamin B-12 (cobalamin) is a complex molecule in which a cobalt atom is contained in a corrin ring. Vitamin B-12 is available in animal protein.
Body stores
Total body stores are 2-5 mg, of which half is stored in the liver. The recommended daily intake is 2 mcg/d in adults; pregnant and lactating women require 2.6 mcg/d. Children require 0.7 mcg/d and, in adolescence, 2 mcg/d. Because vitamin B-12 is highly conserved through the enterohepatic circulation, cobalamin deficiency from malabsorption develops after 2-5 years and deficiency from dietary inadequacy in vegetarians develops after 10-20 years. Its causes are mainly nutritional and malabsorptive, PA being most common.
Physiology of absorption
After ingestion, the low stomach pH cleaves cobalamin from other dietary protein. [11] The free cobalamin binds to gastric R binder, a glycoprotein in saliva, and the complex travels to the duodenum and jejunum, where pancreatic peptidases digest the complex and release cobalamin. Free cobalamin can then bind with gastric intrinsic factor (IF), a 50-kd glycoprotein produced by the gastric parietal cells, the secretion of which parallels that of hydrochloric acid. Hence, in states of achlorhydria, IF secretion is reduced, leading to cobalamin deficiency. Importantly, only 99% of ingested cobalamin requires IF for absorption. Up to 1% of free cobalamin is absorbed passively in the terminal ileum. This why oral replacement with large vitamin B-12 doses is appropriate for PA.
Once bound with IF, vitamin B-12 is resistant to further digestion. The complex travels to the distal ileum and binds to a specific mucosal brush border receptor, cublin, which facilitates the internalization of cobalamin-IF complex in an energy-dependent process. Once internalized, IF is removed and cobalamin is transferred to other transport proteins, transcobalamin I, II, and III (TCI, TCII, TCIII). Eighty percent of cobalamin is bound to TCI/III, whose role in cobalamin metabolism is unknown. The other 20% binds with TCII, the physiologic transport protein produced by endothelial cells. Its half-life is 6-9 min, thus delivery to target tissues is rapid.
The cobalamin-TCII complex is secreted into the portal blood where it is taken up mainly in the liver and bone marrow as well as other tissues. Once in the cytoplasm, cobalamin is liberated from the complex by lysosomal degradation. An enzyme-mediated reduction of the cobalt occurs by cytoplasmic methylation to form methylcobalamin or by mitochondrial adenosylation to form adenosylcobalamin, the 2 metabolically active forms of cobalamin.
Vitamin B-12 role in bone marrow function
In the cytoplasm, methylcobalamin (see image below) serves as cofactor for methionine synthesis by allowing transfer of a methyl group from 5-methyl-tetrahydrofolate (5-methyl-THF) to homocysteine (HC), forming methionine and demethylated tetrahydrofolate (THF). This results in reduction in serum homocysteine, which appears to be toxic to endothelial cells. Methionine is further metabolized to S-adenosylmethionine (SAM).
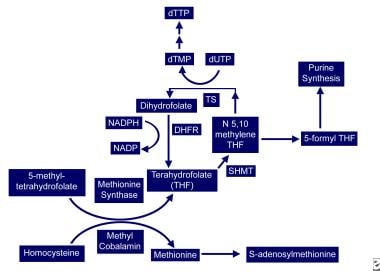 Vitamin B-12–associated neurological diseases. Cobalamin and folate metabolism. TS = thymidylate synthase, DHFR = dihydrofolate reductase, SHMT = serine methyl-transferase.
Vitamin B-12–associated neurological diseases. Cobalamin and folate metabolism. TS = thymidylate synthase, DHFR = dihydrofolate reductase, SHMT = serine methyl-transferase.
THF is used for DNA synthesis. After conversion to its polyglutamate form, THF participates in purine synthesis and the conversion of deoxyuridylate (dUTP) to deoxythymidine monophosphate (dTMP), which is then phosphorylated to deoxythymidine triphosphate (dTTP). dTTP is required for DNA synthesis; therefore, in vitamin B-12 deficiency, formation of dTTP and accumulation of 5-methyl-THF is inadequate, trapping folate in its unusable form and leading to retarded DNA synthesis. RNA contains dUTP (deoxyuracil triphosphate) instead of dTTP, allowing for protein synthesis to proceed uninterrupted and resulting in macrocytosis and cytonuclear dissociation.
Because folate deficiency causes macrocytosis and cytonuclear dissociation via the same mechanisms, both deficiencies lead to megaloblastic anemia and disordered maturation in granulocytic lineages; therefore, folate supplementation can reverse the hematologic abnormalities of vitamin B-12 deficiency but has no impact on the neurologic abnormalities of vitamin B-12 deficiency, indicating both result from different mechanisms.
Vitamin B-12 role in the peripheral and central nervous systems
The neurologic manifestation of cobalamin deficiency is less well understood. CNS demyelination may play a role, but how cobalamin deficiency leads to demyelination remains unclear. Reduced SAM or elevated methylmalonic acid (MMA) may be involved.
SAM is required as the methyl donor in polyamine synthesis and transmethylation reactions. Methylation reactions are needed for myelin maintenance and synthesis. SAM deficiency results in abnormal methylated phospholipids such as phosphatidylcholine, and it is linked to central myelin defects and abnormal neuronal conduction, which may account for the encephalopathy and myelopathy. In addition, SAM influences serotonin, norepinephrine, and dopamine synthesis. This suggests that, in addition to structural consequences of vitamin B-12 deficiency, functional effects on neurotransmitter synthesis that may be relevant to mental status changes may occur. Parenthetically, SAM is being studied as a potential antidepressant.
Another possible cause of neurologic manifestations involves the other metabolically active form of cobalamin, adenosylcobalamin (see image below), a mitochondrial cofactor in the conversion of L-methylmalonyl CoA to succinyl CoA. Vitamin B-12 deficiency leads to an increase in L-methylmalonyl-CoA, which is converted to D-methylmalonyl CoA and hydrolyzed to MMA. Elevated MMA results in abnormal odd chain and branched chain fatty acids with subsequent abnormal myelination, possibly leading to defective nerve transmission.
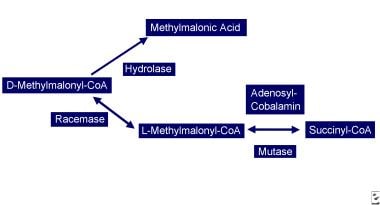 Vitamin B-12–associated neurological diseases. Cobalamin deficiency leads to reduced adenosylcobalamin, which is required for production of succinyl-CoA. D-methylmalonyl-CoA is converted to methylmalonic acid.
Vitamin B-12–associated neurological diseases. Cobalamin deficiency leads to reduced adenosylcobalamin, which is required for production of succinyl-CoA. D-methylmalonyl-CoA is converted to methylmalonic acid.
More recent studies propose a very different paradigm: B-12 and its deficiency impact a network of cytokines and growth factors, ie, brain, spinal cord, and CSF TNF-alpha; nerve growth factor (NGF), IL-6 and epidermal growth factor (EGF), some of which are neurotrophic, others neurotoxic. Vitamin B-12 regulates IL-6 levels in rodent CSF. In rodent models of B-12 deficiency parenteral EGF or anti-NGF antibody injection prevents, like B-12 itself, the SCD-like lesions.
In the same models, the mRNAs of several cell-type specific proteins (glial fibrillary acidic protein, myelin basic protein) are decreased in a region specific manner in the CNS, but, in the PNS myelin, protein zero and peripheral myelin protein 22 mRNA remain unaltered.
In human and rodent serum and CSF, concomitantly with a vitamin B-12 decrease, EGF levels are decreased, while at the same time, TNF-alpha increases in step with homocysteine levels. These observations provide evidence that the clinical and histological changes of vitamin B-12 deficiency may result from up-regulation of neurotoxic cytokines and down-regulation of neurotrophic factors. [12]
N
N2 O can oxidize the cobalt core of vitamin B-12 from a 1+ to 3+ valance state, rendering methylcobalamin inactive, inhibiting HC conversion to methionine and depleting the supply of SAM. Patients with sufficient vitamin B-12 body stores can maintain cellular functions after N2 O exposure, but in patients with borderline or low vitamin B-12 stores, this oxidation may be sufficient to precipitate clinical manifestations.
Epidemiology
Frequency
The prevalence of vitamin B-12 deficiency is difficult to ascertain because of diverse etiologies and different assays (i.e., radioassay or chemiluminescence). Affected individuals may number 300,000 to 3 million in the United States.
Using the radioassay and a value less than 200 pg/mL, the prevalence of vitamin B-12 deficiency is 3-16%. In a geriatric population using a radioassay cutoff of 300 pg/mL and elevated HC and MMA levels, a prevalence of 21% was reported.
Of HIV-seropositive individuals, 11% are vitamin B-12 deficient; another 12% have levels of 200-240 pg/mL. In a subgroup with chronic diarrhea, the rate reaches 39%. However, the importance for vitamin B-12 deficiency in the development of neurologic disease in these patients remains unclear.
In Europe, the prevalence of vitamin B-12 deficiency is 1.6-10%.
In India, a hospital population radioassay study with a cutoff of 200 pg/mL found a vitamin B-12 deficiency in 0.88% of patients, with borderline values in 3.8%.
Mortality/Morbidity
Vitamin B-12 deficiency is associated with an elevated HC.
The prevalence of hyperhomocysteinemia in the general population is 5-10%; in people older than 65 years it may reach 30-40%. Elevated HC is a risk factor for coronary artery, cerebrovascular, and peripheral vascular diseases and venous thrombosis. About 10% of the vascular disease risk in the general population is linked to HC.
Case-control studies have reported a correlation between multi-infarct dementia or dementia of the Alzheimer type and elevated HC; vitamin B-12 supplementation had no clinical benefit.
Neural tube defects are associated with low folate and vitamin B-12.
PA patients have a 3 times and 13 times increased risk of gastric carcinoma and gastric carcinoid tumors, respectively.
Patients with diabetes mellitus type 1 and autoimmune thyroid diseases are at higher risk of developing PA. A 2009 study noted a 22% prevalence of vitamin B-12 deficiency in patients with diabetes mellitus type 2. [13]
Multifactorial abnormalities of vitamin B-12 metabolism and absorption occur in HIV infection.
Race
PA prevalence may be higher in white people and lower in Hispanic and black people.
No known relationship exists between neurologic symptoms and race.
Studies in Africa and the United States have shown higher vitamin B-12 and transcobalamin II levels in black than in white individuals. Additionally, blacks have lower HC levels and metabolize it more efficiently than whites.
Sex
In Europe and Africa, the prevalence of PA is higher in elderly women than men (1.5:1), while in the United States no differences exist.
Men have higher HC levels at all ages.
Pregnancy and estrogen replacement in postmenopausal women lower HC levels.
Age
PA occurs in people of all ages, but it is more common in people older than 40-70 years and, in particular, in people older than 65 years.
In white people, the mean age of onset is 60; in black people, the mean age is 50 years.
Congenital PA manifests in children aged 9 months to 10 years; the mean age is 2 years.
Prognosis
Therapy with vitamin B-12 in subacute combined degeneration stops progression and improves neurologic deficit in most patients.
Younger patients with less severe disease and short duration illness do better.
In a large retrospective review of 57 patients with subacute combined degeneration, absence of sensory level, absent Rhomberg sign, and flexor planter reflex were associated with good prognosis. [29]
On spinal MRI, involvement of less than 7 spinal segments, cord swelling, and enhancement, but not cord atrophy, were associated with better prognosis. [29]
Clinical improvement is most pronounced in the first 2 months but continues up to 6 months.
-
Vitamin B-12–associated neurological diseases. Cobalamin and folate metabolism. TS = thymidylate synthase, DHFR = dihydrofolate reductase, SHMT = serine methyl-transferase.
-
Vitamin B-12–associated neurological diseases. Cobalamin deficiency leads to reduced adenosylcobalamin, which is required for production of succinyl-CoA. D-methylmalonyl-CoA is converted to methylmalonic acid.
-
Vitamin B-12–associated neurological diseases. Pernicious anemia. Characteristic lemon-yellow pallor with raw beef tongue lacking filiform papillae. Photo from Forbes and Jackson with permission.
-
Vitamin B-12–associated neurological diseases. Fluid attenuated inversion recovery (Flair) MRI sequence in a patient with cobalamin deficiency and neuropsychiatric manifestations. Discrete areas of hyperintensities are present in the corona radiata.





