Practice Essentials
Tuberous sclerosis complex (TSC) is a genetic disorder affecting cellular differentiation, proliferation, and migration early in development, resulting in a variety of hamartomatous lesions that may affect virtually every organ system of the body.
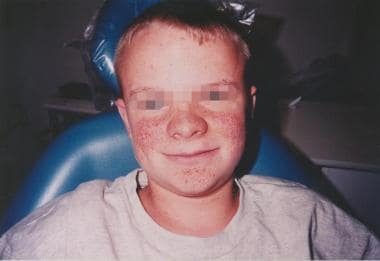
The best-known cutaneous manifestation of TSC is adenoma sebaceum, which often does not appear until late childhood or early adolescence. This lesion is an angiofibroma (ie, cutaneous hamartoma) and is not related to excessive sebum or acne. Flat, reddish macular lesions develop first, which can be mistaken for freckles early on. See the image below.
Signs and symptoms
Findings in TSC include the following:
-
Neurologic findings: Abnormal neurologic findings result from the location, size, and growth of tubers and the presence of subependymal nodules (SENs) and subependymal giant cell astrocytomas (SEGAs)
-
Cutaneous findings: The best-known cutaneous manifestation of TSC is adenoma sebaceum, which often does not appear until late childhood or early adolescence
-
Cardiac findings: Cardiac involvement is usually maximal at birth or early in life; it may be the presenting sign of TSC, particularly in early infancy; 50-60% of individuals with TSC have evidence of cardiac disease, mostly rhabdomyomas.
-
Ophthalmic findings: At least 50% of patients have ocular abnormalities; some studies have reported a prevalence as high as 80%; these lesions are actually retinal astrocytomas that tend to become calcified over time
-
Pulmonary findings: Prospective and retrospective studies have found cystic pulmonary abnormalities in as many as 40% of women with TSC
-
Renal findings: Renal manifestations of TSC are the second most common clinical feature; 4 types of lesions can occur: autosomal dominant polycystic kidney disease lesions, isolated renal cyst(s), angiomyolipomas (AMLs), and renal cell carcinomas
-
Dental findings: Pitting of the dental enamel is invariably present in the permanent teeth of patients with TSC [1] ; gingival fibromas occur in 70% of adults with TSC, in 50% of children with mixed dentition (primary and permanent teeth), and in 3% of children with only primary teeth
-
Gastrointestinal findings: Hamartomas and polyposis of the stomach, intestine, and colon may occur
-
Hepatic findings: Hepatic cysts and hepatic AMLs, typically asymptomatic and nonprogressive, have been reported in as many as 24% of patients with TSC, with a marked female predominance (female-to-male ratio 5:1)
-
Skeletal findings: Sclerotic and hypertrophic lesions of bone may be found incidentally on radiography performed for other indications
See Clinical Presentation for more detail.
Diagnosis
Laboratory studies
Laboratory studies are performed as indicated clinically to identify genetic mutations associated with TSC, monitor anticonvulsant treatment, identify idiosyncratic or dose-related adverse effects, and identify or monitor underlying renal or pulmonary disease. Diagnosis should be possible in most cases using established clinical criteria. Molecular genetic testing is useful in uncertain or questionable cases, as well as for prenatal diagnosis and for screening family members of an affected individual.
Imaging studies
The following 3 imaging studies are usually undertaken in patients with TSC:
-
Computed tomography scanning or magnetic resonance imaging of the brain: Performed to identify SEGAs before obstructive hydrocephalus occurs; they also identify the extent and number of cortical tubers present [2]
-
Renal ultrasonography: Performed to assess change in AMLs or cysts, in the hope that this will allow operative intervention prior to the development of renal failure
-
Echocardiography: Performed as part of the baseline evaluation in a patient with newly diagnosed or suspected TSC
Additional tests
Other tests used in the assessment of patients with TSC include the following:
-
Electroencephalography: Should be performed in patients with TSC in whom seizures are suspected; follow-up electroencephalography is performed as clinically indicated
-
Electrocardiography: Baseline electrocardiography is recommended for all patients newly diagnosed with TSC, since cardiac arrhythmias, although rare, may have sudden death as their presenting symptom
See Workup for more detail.
Management
Pharmacologic treatment
Antiepileptic medications (AEDs) are the mainstay of therapy for patients with TSC. The choice of specific AED(s) for treating seizures in patients with TSC is based on the patient's seizure type(s), epilepsy syndrome(s), other involved organ systems, and age, along with AED side-effect profiles and available formulations.
The drug everolimus (Afinitor) helped to reduce kidney tumors linked to tuberous sclerosis complex (TSC) in a double-blind, placebo-controlled, phase-3 trial. The efficacy of the drug, which has been approved for use against TSC in the United States and Europe, was measured by the proportion of patients (diagnosed with tuberous sclerosis or sporadic lymphangioleiomyomatosis) in whom target angiolipomas were reduced by at least half of their total volume relative to baseline. [3, 4]
In the study, 118 patients (median age 31) from 24 centers in 11 countries received either everolimus (n=79) or placebo (n=39). Angiomyolipomas had a 42% response rate to everolimus and a 0% response rate to placebo.
In addition, everolimus has been shown to significantly reduce seizure frequency, with 28.2% of patients receiving low exposure/low dosage demonstrating 50% or greater decrease in seizures, and 40% of patients receiving high exposure/high dosage demonstrating 50% or greater decrease in seizures. [5]
Surgery
Surgical treatment of patients with TSC can include the following:
-
Focal cortical resection/thermal ablation
-
Corpus callosotomy
-
Vagus nerve stimulation
In addition, SEGAs require resection if they produce hydrocephalus or significant mass effect. If a gross total resection can be achieved, recurrence is unlikely.
See Treatment and Medication for more detail.
Background
In 1880, Bourneville first described the cerebral manifestations of this disorder, applying the term "sclerose tubereuse" to indicate the superficial resemblance of the lesions to a potato. In 1908 Vogt set forth the triad of intractable epilepsy, intellectual disability, and adenoma sebaceum; this description (until relatively recently) represented the hallmark of tuberous sclerosis complex (TSC) to most clinicians. Unfortunately, this concept led many primary care physicians and even neurologists to conclude, incorrectly, that a diagnosis of TSC predestines a child to crippling, lifelong neurological and psychological morbidity.
TSC is now known to be a genetic disorder affecting cellular differentiation, proliferation, and migration early in development, resulting in a variety of hamartomatous lesions that may affect virtually every organ system of the body. Less than one third of affected persons fit the classic Vogt triad.
Pathophysiology
Clinically, tuberous sclerosis complex (TSC) exhibits an autosomal dominant inheritance pattern, with a high spontaneous mutation rate. Two distinct genetic loci responsible for TSC have been identified: one on chromosome band 9q34 (also referred to as TSC1) and another on chromosome band 16p13 (TSC2).
The TSC2 gene was identified in 1993, and its protein product has been named tuberin. Tuberin has GTPase-activating properties and seems to function as a tumor suppressor. The highest levels of tuberin are found in adult human brain, heart, and kidney; tuberin also has been localized to arterioles of kidney, skin, and heart, as well as to pyramidal neurons and cerebellar Purkinje cells. Its exact function, particularly during neurogenesis, remains unknown. Individual tubers are thought to arise developmentally when mutated neural progenitor cells in the subependymal germinal matrix give rise to abnormally migrating daughter cells that in turn produce tubers. The tubers may undergo cystic degeneration or calcification, or exhibit contrast enhancement on neuroimaging, but these features do not necessarily imply malignant transformation.
Hamartin, the TSC1 product, was identified in 1997 and may also function as a tumor suppressor. Rather than having completely separate functions, both hamartin and tuberin have been shown to have "coiled-coil" domains that interact with each other. [6] Hamartin and tuberin together form a tumor suppressor complex, which, through the GTPase activating function of tuberin, drives the small GTPase (termed Ras, homolog enhanced in brain) or Rheb into the inactive GDP-bound state. Rheb in the GTP-bound, active state is a positive effector of mTOR [7] (mTOR, m ammalian t arget o f r apamycin—so named because of its ability to bind to the immunosuppressant drug rapamycin [sirolimus, Rapamune] before its function was known) (see following image).
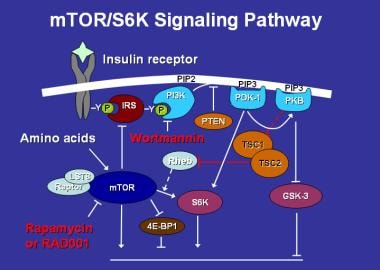 Mammalian target of rapamycin (mTOR) activates the protein S6 kinase, which enhances cell growth and protein synthesis. It, in turn, is regulated by multiple factors, including insulin, amino acids, the drugs rapamycin and its congeners (eg, RAD001), and the TSC gene products via the GTPase-activating protein Rheb.
Mammalian target of rapamycin (mTOR) activates the protein S6 kinase, which enhances cell growth and protein synthesis. It, in turn, is regulated by multiple factors, including insulin, amino acids, the drugs rapamycin and its congeners (eg, RAD001), and the TSC gene products via the GTPase-activating protein Rheb.
mTOR, a major effector of cell growth (as opposed to cell proliferation) functions, among other things, as a sort of master switch for cellular anabolism versus catabolism, and it has important regulatory functions for cell volume and protein synthesis. It is also regulated by a wide variety of other factors, including insulin and amino acids. mTOR is a highly conserved protein kinase in evolution and is present in a wide range of organisms, from yeast, to Drosophila, to mammals.
Mutations in either hamartin or tuberin drive Rheb into the GTP-bound state, which results in constitutive mTOR signaling. mTOR appears to mediate many of its effects on cell growth through the phosphorylation of the ribosomal protein S6 kinases (S6Ks) and the repressors of protein synthesis initiation factor eIF4E, the 4EBPs. The S6Ks act to increase cell growth and protein synthesis, whereas the 4EBPs serve to inhibit these processes. mTOR interacts with the S6Ks and the 4EBPs through an associated protein, Raptor. When mTOR is constitutively activated through mutations in either hamartin or tuberin this results in the hamartomatous lesions of tuberous sclerosis in the brain, kidneys, heart, lungs, and other organs.
Rapamycin is capable of inducing regression of renal angiomyolipomas in animal models of TSC, and this effect appears to be enhanced by interferon-gamma, whose receptors are up-regulated by overactivity of mTOR. This pathway may be excessively active in other human malignancies as well as in TSC. These observations raise the possibility of new therapeutic interventions for this disorder. Trials of rapamycin for renal angiomyolipomas in humans with TSC have been completed (see Treatment section). Multicenter, randomized, placebo-controlled studies investigating RAD001 (everolimus) in the treatment of angiomyolipomatas (AMLs) and subependymal giant cell astrocytomas (SEGAs) are currently underway. On November 1, 2010, everolimus was approved by the US Food and Drug Administration (FDA) for SEGAs associated with tuberous sclerosis that cannot be treated with surgery.
The high incidence of sporadic TSC, coupled with a probable "second hit" phenomenon, seems a likely explanation for the marked phenotypic variability observed. The second hit hypothesis suggests that in addition to an inherited or sporadic autosomal mutation in one allele of either TSC 1 or TSC 2, clinical signs and/or symptoms manifest only after a further mutation or inactivating event in the second, unaffected allele (“second hit”). This allows considerable potential for diversity, not only among various deletions and mutations between 2 genetic loci, but also with regard to possible interactions between protein products of varying functionality arising from different mutations on each allele. Thereby adjacent tubers, angiomyolipomas, even facial angiofibromas can have different second hits and different genotypes within the same organ of the same patient.
Further complicating the high spontaneous mutation rate is the observation that parents of an affected child, who themselves show no sign of TSC, nonetheless have an increased risk (approximately 2% overall) of having additional affected children. This is thought to result from parental mosaicism for one of the TSC genes limited to cells of their germ line (ie, gonadal tissues). True failure of penetrance of the TSC genes is believed to be rare.
Recent research has identified phenotypic differences as they may relate to particular genotypes. Linkage studies initially suggested a roughly equal distribution of TSC1 and TSC2 mutations among affected individuals. However, subsequent mutational analysis has shown TSC2 mutations to be present in 80-90% of affected individuals, while TSC1 mutations are present in 10-20%. The TSC2 gene is contiguous with the gene producing polycystic kidney disease (PKD1). Individuals with features of both TSC and polycystic kidney disease (as opposed to simple renal cysts) likely have deletions spanning both genes.
Jones et al found a higher incidence of "mental handicap" in persons with TSC2 mutations than in those with TSC1 mutations. They identified mental handicap retrospectively in relatively broad terms: developmental quotient less than 70, inability to attend regular school without supplementary assistance, institutionalization, requiring assistance with daily activities, etc.
Dabora et al recently described genotypic and phenotypic features in 224 persons with TSC. [8] A TSC2 genetic abnormality was found to be associated consistently with more severe clinical disease regardless of organ system. Although prominent phenotypic variability was still the rule, patients with TSC2 abnormalities were more apt to have higher tuber counts, refractory seizures, autism, larger AML and/or cardiac rhabdomyomata, and more severe cutaneous lesions. This suggests that, while tuberin and hamartin have similar functions, tuberin plays a more critical role in regulation of cellular differentiation. While TSC2 mutations are more apt to be associated with severe clinical phenotypes, they predominate in all forms of the disease, mild and severe, familial and sporadic. Spontaneous mutations are also much more likely to reflect TSC2 disease. Suggestions that TSC1 disease is more likely familial than sporadic appear to be incorrect.
Epidemiology
Frequency
Birth incidence of tuberous sclerosis complex (TSC) is 1 case per 6,000 population, with a prevalence of 1 case per 10,000 population.
Factors that hamper accurate assessment of incidence and prevalence include under-recognition of less severe phenotypes, high spontaneous mutation rate (approximately two thirds), marked variability of symptoms (even within specific kindreds of affected individuals), and reluctance of asymptomatic parents and relatives to undergo diagnostic testing related to concerns of uninsurability and social stigma.
Mortality/Morbidity
Complications of neurological involvement are the most common causes of mortality and morbidity. These are due chiefly to intractable epilepsy, status epilepticus, and subependymal giant cell astrocytoma (SEGA) with associated hydrocephalus.
Renal complications are the next most frequent cause of morbidity and death. These usually arise from an enlarging AML, resulting in retroperitoneal hemorrhage. End-stage renal disease can occur, as a result of either destruction of normal renal parenchyma by an enlarging AML or polycystic kidney disease.
Less common are cardiac arrhythmias (which can present with sudden, unexplained death), congestive heart failure, and end-stage lung disease. Cardiac involvement is maximal in infants and exhibits spontaneous regression as the child grows older. Pulmonary disease occurs predominantly in women in the third and fourth decades of life.
Race-, sex-, and age-related statistics
TSC affects all races without a clear-cut predominance.
TSC affects both sexes equally. Some studies have suggested that males are more likely to suffer neurological morbidity, but this has not been demonstrated conclusively.
TSC can present at any age. In infants and children, it usually is identified as a cause of epilepsy, autism, or cardiac failure.
Older persons [9] may present with renal failure or pulmonary or cutaneous manifestations in the absence of prominent, or any, neurological symptoms.
Various organ systems are affected maximally at different points in life.
-
Cardiac involvement occurs during the intrauterine or neonatal period.
-
Rhabdomyomas tend to regress over time.
-
Epilepsy, autism, and developmental delays manifest themselves from infancy to adolescence.
-
Polycystic kidney disease usually is apparent in infancy or early childhood.
-
AMLs may develop at any time from childhood into adult life.
-
Lymphangiomyomatosis typically presents in the third or fourth decade of life.
Prognosis
The prognosis of patients with tuberous scelrois complex (TSC) is not as grim as has been typically thought. Higher numbers of tubers, earlier onset and intractability of seizures, and infantile spasms are associated with (but do not guarantee) worse cognitive and behavioral outcomes (see images below). Cardiac lesions almost always spontaneously regress, although supportive care may be necessary for a time. Pulmonary and renal lesions affect prognosis on the basis of their extent and severity.
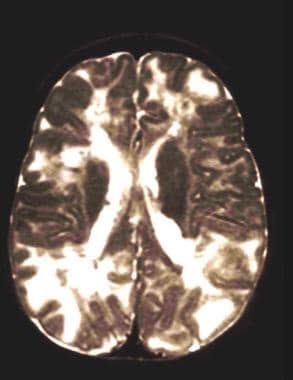 Multiple tubers in a child with tuberous sclerosis, normal intelligence, and well-controlled seizures. High tuber count does not invariably mean poor neurological outcome.
Multiple tubers in a child with tuberous sclerosis, normal intelligence, and well-controlled seizures. High tuber count does not invariably mean poor neurological outcome.
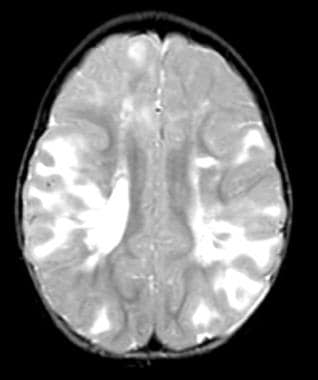 All tubers are not equal. This child has a smaller number of tubers than the patient shown in the previous image, but the tubers are larger in size. She too has normal intelligence and is seizure free on medication.
All tubers are not equal. This child has a smaller number of tubers than the patient shown in the previous image, but the tubers are larger in size. She too has normal intelligence and is seizure free on medication.
-
Enhancing subependymal nodules, including a probable giant cell astrocytoma in the region of the foramen of Monro. Subependymal nodules may increase in size over time from one scan to the next, and then stabilize. This lesion had not changed with serial imaging over 2 years. The patient remains asymptomatic and is monitored closely for any deterioration.
-
Hydrocephalus from a subependymal giant cell astrocytoma in a patient with tuberous sclerosis. The patient presented with acute blindness and ataxia.
-
Facial angiofibromas in a young man with tuberous sclerosis complex.
-
Dysplastic periungual fibroma involving the great toe in a patient with tuberous sclerosis.
-
Gingival fibromas (see arrows) in a patient with tuberous sclerosis. A stain outlines dental pits and craters. Gingival hyperplasia from other causes (eg, phenytoin use) is more diffuse and usually not nodular/focal in nature.
-
Typical ash leaf macules; the reddish, nodular area at the upper lumbar area is a shagreen patch.
-
Atrial rhabdomyoma as seen on cardiac CT scan in a patient with tuberous sclerosis.
-
Nonobstructive ventricular rhabdomyomas in a patient with tuberous sclerosis.
-
Ventricular rhabdomyomas may diffusely infiltrate the myocardium, as in this patient with tuberous sclerosis. The patient presented with cardiac failure and hydrops at birth. After a period of intensive supportive care and inotropic therapy, she now has essentially normal cardiac function and is on no medications.
-
Multifocal pulmonary cysts characteristic of lymphangiomyomatosis. As many as 40% of women with tuberous sclerosis have pulmonary cysts on chest CT scan.
-
Massive bilateral angiomyolipomas in a woman with tuberous sclerosis. She also has lymphangiomyomatosis.
-
Pre-embolization angiography of the patient with angiomyolipomas shown the previous image. Dysplastic arterial vessels are demonstrated.
-
Vessels to the angiomyolipoma shown in the previous image have been occluded with coils. This should produce regression of the lesion and prevention of hemorrhage. Functional intervening renal parenchyma is preserved.
-
Enamel pitting in tuberous sclerosis. Pinpoint size pitting (A) and crater size pitting (B) are visible. Red dye is used to enhance recognition.
-
Basilar artery aneurysm in a 2-year-old girl with tuberous sclerosis. The arrow shows the anterior aspect of the aneurysm where it abuts the clivus. The lesion was not present on MRI performed 11 months earlier.
-
This presumed tuber was first noted in the left frontal region. It expanded in size, affecting adjacent structures across the midline and resulting in calcifications still evident in the right frontal region. The tuber then spontaneously involuted. About 20% of tubers may show changes in imaging characteristics over time, requiring close imaging follow-up. This patient remained asymptomatic from the mass effect, and his seizures resolved as the lesion involuted.
-
This father and all 3 children have tuberous sclerosis complex. The children are now grown up and of normal intelligence, including the young lady at left who is cushingoid from therapy with adrenocorticotropic hormone for infantile spasms.
-
The child whose CT scan is shown presented with medically intractable epilepsy thought to be due to partial hemimegalencephaly. She became seizure free after partial hemispherectomy. Pathology was consistent with a cortical tuber. She was subsequently found to have multiple ash leaf macules and diagnosed with tuberous sclerosis.
-
Multiple tubers in a child with tuberous sclerosis, normal intelligence, and well-controlled seizures. High tuber count does not invariably mean poor neurological outcome.
-
All tubers are not equal. This child has a smaller number of tubers than the patient shown in the previous image, but the tubers are larger in size. She too has normal intelligence and is seizure free on medication.
-
Mammalian target of rapamycin (mTOR) activates the protein S6 kinase, which enhances cell growth and protein synthesis. It, in turn, is regulated by multiple factors, including insulin, amino acids, the drugs rapamycin and its congeners (eg, RAD001), and the TSC gene products via the GTPase-activating protein Rheb.
-
Subependymal giant cell astrocytoma prior to stereotactic insertion of balloon catheter as seen on T2-weighted MRI.
-
Modified angioplasty catheter used in creation of surgical tract for astrocytoma resection.
-
Catheter placed in proximity to lesion, balloon inflated.
-
Postoperative T2-weighted MRI in a patient with subependymal giant cell astrocytoma showing gross total resection of giant cell astrocytoma with minimal disruption of overlying cortex.
-
Mean reduction in simple and complex partial seizures in patients with tuberous sclerosis complex (TSC) who were treated with vagus nerve stimulator at the author's institution at 6 and 12 months. Overall reduction in secondarily generalized seizures was 22% at 12 months (N = 17; 10 boys, 7 girls, aged 3-12 y).
-
Regression of a giant cell astrocytoma after approximately 15 months oral rapamycin therapy in a 4-year-old patient with tuberous sclerosis.






