Overview
Due to the efforts of numerous clinicians and basic scientists over many years, we now have a firm understanding of how the motor signal generated in the brain travels down the spinal cord, into a peripheral nerve, and interfaces with the target muscle. Furthermore, we are now able to analyze many aspects of this complex process in the clinic or at the bedside via clinical examination findings, pharmacological challenge, or electrophysiological equipment (see the image below). These advances in scientific understanding that have directly impacted clinical practice represent an excellent example of translational research.
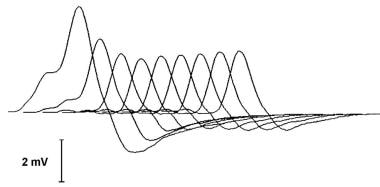 A typical decrementing response to repetitive nerve stimulation in myasthenia gravis. The amplitude of the initial response is normal, and the decrement is maximal in the fourth response. Thereafter, the responses increase somewhat, giving a U-shaped envelope to the train of responses.
A typical decrementing response to repetitive nerve stimulation in myasthenia gravis. The amplitude of the initial response is normal, and the decrement is maximal in the fourth response. Thereafter, the responses increase somewhat, giving a U-shaped envelope to the train of responses.
When discussing diseases of the neuromuscular junction (NMJ), many physicians first think of myasthenia gravis (MG). This is entirely appropriate, as research into the pathogenesis of MG has always been at the center of our understanding of the NMJ. The first patient with MG described in medical writings was likely a Native American named Chief Opechankanough, uncle of the famous Matoaka (Pocahontas). Colonial physicians who examined him in the early 1600s documented weakness and ptosis that improved with rest. Their accounts were not formally published in a medical journal. [1, 2] However, in 1672, Thomas Willis (for whom the Circle of Willis is named) published the case of an English woman whose clinical findings seemed consistent with MG. [3]
Numerous cases were subsequently described. Several accounts were documented by William Erb in 1879, and in 1893, Samuel Goldflam provided excellent clinical descriptions. Friedrich Jolly coined the term myasthenia gravis pseudoparalytica in 1895 and also pioneered one of the assessment methods, repetitive stimulation, which is discussed later. [4]
The next advance occurred in the 1930s when English doctor Mary Broadfoot Walker, one of the first female British physicians, showed that injections of physostigmine could temporarily increase the strength of individuals with MG and reverse symptoms such as ptosis. She reported these findings in Lancet in 1934. The Tensilon test, discussed later, is the modern version of Dr. Walker's technique. Her work, particularly when combined with the basic pharmacologic studies of contemporaries such as Dale and Feldberg on cholinergic neuromuscular transmission, suggested impairment of cholinergic transmission at the nerve-muscle synapse. [5]
Approximately 30 years prior to Dr. Walker's findings, more insight into to pathophysiology of MG had emerged. In 1901, the famous pathologist Karl Weigert noted that lymphoid cells were present in the muscle and other tissues of persons with this affliction. He also made the connection between MG and thymic hypertrophy and/or neoplasia. Four years later, neurologist E. Farquhar Buzzard noted collections of lymphocytes (lymphorrhages) in the tissues of patients with MG. Shortly after the observations of Weigert and Buzzard became known, clinicians began to use thymectomy as a treatment for MG. [6] Initially, only actual thymomas were resected, but the practice soon expanded to include thymic hyperplasia and ultimately removal of the thymus for patients without any apparent thymic abnormality. [7, 8]
Throughout the first part of the twentieth century, evidence for the immunological basis of MG continued to accumulate. In 1960, Dr John A. Simpson introduced his hypothesis that the cause of MG was "an 'autoimmune' response of muscle in which an antibody to end-plate protein may be formed." [9]
Nonetheless, many competing hypotheses remained active for the next 15 years. This was largely due to 2 primary points of contention: (1) Which side of the nerve-muscle junction was the defect in MG (the presynaptic nerve endings or the postsynaptic motor end plate)? and (2) What was the fundamental etiology of the disease (eg, autoimmune disease as Simpson had speculated)? [10, 11]
In the 1970s, new techniques emerged that allowed for further investigation of the function of the neuromuscular junction. In particular, the use of alpha-bungarotoxin, a compound derived from snake venom, that bound to specific portions of the acetylcholine receptor (AChR) was pioneered by pharmacologists C. C. Chang, C. Y. Lee, and L. F. Tseng. Further contributions from clinician-scientists such as Andrew Engel, Douglas Fambrough, Edward Lambert, Vanda Lennon, Daniel Drachman, Eric Stalberg, Joze Trontelj, Jan Ekstedt, John Newsom-Davis, David Richman, Marjorie Seybold, and Klaus Toyka helped to integrate the emerging immunologic-neurophysiologic connections suggesting that the disorder was secondary to immunologic dysfunction at the postsynaptic motor end plate.
Jim Patrick, Jon Lindstrom, and associates at the Salk Institute produced antibodies to various subunits of the AChR and ultimately demonstrated that the disease could be reproduced by antibodies to this receptor. [12, 13, 14, 15, 16, 17, 18, 19, 20]
The highly detailed knowledge of neuromuscular transmission that we appreciate today is largely due to the efforts toward understanding MG and other diseases of the NMJ. To be certain, some aspects remain unknown. However, the essential framework is now common knowledge to most students early in medical school and can be summarized as follows:
A nerve action potential arrives at the axon terminal.
Voltage-gated calcium channels open and calcium ions enter the neuron.
A biochemical cascade ensues, causing vesicles containing acetylcholine (ACh) to fuse with the cell membrane, releasing ACh into the synaptic cleft.
ACh diffuses across the synaptic cleft and binds to nicotinic AChRs on the postsynaptic cell membrane.
The AChRs, which are sodium ion channels, open and allow sodium ions to flow into the muscle cell. Several other nearby membrane proteins including agrin, rapsyn, and muscle-specific tyrosine kinase (MuSK) influence both the function and the positioning of the AChRs.
The influx of sodium (accompanied by an exit of potassium through potassium channels) causes depolarization of the muscle cell membrane.
If depolarization is sufficient, it triggers a regenerative muscle action potential that spreads throughout the cell into the transverse tubules. This causes a release of calcium from the sarcoplasmic reticulum, which initiates muscle contraction.
When the ACh molecule is released from the AChR, it is degraded by the enzyme acetylcholinesterase (AChE) into acetate and choline. After reuptake by the neuron, choline is combined with another acetate group to reform acetylcholine, which is subsequently repackaged into a vesicle.
Our current understanding of neuromuscular transmission is now complemented by modern methods for diagnosis of dysfunction at the myoneural synapse. The primary components for clinical assessment are history and physical examination, pharmacologic challenge, neurophysiologic (electrical) studies and neuroimmunologic assays. Each of these are discussed in the following sections.
Myasthenia Gravis: Clinical Evaluation
Myathenia gravis (MG) is described in detail in Myasthenia Gravis. However, key points relevant to neuromuscular transmission are presented here, as it would be impossible to discuss neuromuscular transmssion without discussing MG.
History
A history of isolated ocular sympotoms such as ptosis and diplopia occur in ocular myasthenia, while generalized myasthenia (MGg) presents with additional symptoms such as extremity weakness, swallowing difficulty, and respiratory muscle weakness. These symptoms are, of course, present in other entities.
A hallmark of MGg is fatigability of the muscle, which typically worsens toward the end of the day. However, rather than asking about fatigue and/or weakness late in the day, it is often more helpful to inquire whether fatigue is lessened by resting for a few minutes. Temporary restoration of energy after rest is often more specific than afternoon or evening exhaustion. An excellent question to pose to the patient would be "If you rest for a few minutes, does that temporarily restore your strength?" However, this is not very specific. [21]
Physical examination
The physical examination includes a complete neuromuscular exam with specific focus on the the problems elicited during the history.
Fatigability can be tested, at least to an extent, by asking the patient to perform repetitive motions or to hold fixed postures. The examiners can use themselves as a control.
Eyelid function, especially the ability to keep the eyes open is easy to observe, and is useful to test if MG is suspected. Since ptosis is such a common sign in MG, several diagnostic methods are available that use ptosis as an index of impaired neuromuscular transmission. Knowing some of these relatively easy physical diagnostic methods can be useful. None of these are specific for MG or for any type of neuromuscular transmission, but in the proper setting they can be quite helpful.
The Simpson test is the simplest. This requires the patient to look up at a fixed point for an extended period, which frequently brings out the ptosis in a patient with MG, that is, preexisting ptosis might worsen after this maneuver or one may see ptosis that was not initially present.
Another quick test is to examine for the Cogan lid twitch sign. In this test, the patient looks down for 10-15 seconds and then returns to primary gaze. A brief twitch indicates a positive sign on the affected side. One can also have the patient look down for 15 seconds, then look up before returning to primary gaze to elicit the twitch. [22, 23]
Another test, the extended fatigue and recovery bedside test, was published by Toyka. [24] This test involves instructing the patient to close both lids with maximal contraction for 10-30 seconds, then open both eyes. This leads to temporary improvement in preexisting ptosis in the affected eyelid. Also, the Cogan lid twitch sign may be seen occasionally when opening the eyes.
The aforementioned tests are essentially dependent upon the fatigability of the neuromuscular junction. The test for enhanced ptosis brings the central nervous system into play. If a patient has ptosis in one eye but not the other eye, simply hold the ptotic eye open (ie, pull the lid up further than the patient can do on their own). Frequently, this will cause ptosis to appear in the other eye. The reason is supposedly that the CNS is directing more effort to keeping the ptotic eye open. However, according to the Herring law, the CNS distributes the impetus to both eyes. [25] Thus, by helping the patient keep the ptotic eye open, the CNS is tricked into reducing the effort and this brings out the ptosis in the other eye. [26]
Ice pack test
The ice pack test can be viewed as a transition between the clinical methods listed above and the more complicated studies discussed later. For the ice pack test, an ice pack is applied to the ptotic eye(s). A positive test result is when one observes improvement in the ptosis shortly after the application of the ice. This is because neuromuscular transmission improves at cooler temperatures. [27] This test is generally believed to have good sensitivity and specificity, though it is probably more subject to false-positive and false-negative results than the Tensilon test. Holding an ice pack over the eye for 1-2 minutes is unlikely to cause harm. [28, 29, 30]
Tensilon test
Edrophonium chloride (Enlon, Reversol, Tensilon) is primarily used as a diagnostic tool to predict the response to longer-acting cholinesterase inhibitors. As with other cholinesterase inhibitors, edrophonium chloride decreases the metabolism of acetylcholine, increasing the cholinergic effect at the myoneural junction.
Weakness improves after intravenous administration of Tensilon. For a Tensilon test result to be considered positive, a dramatic, unequivocal improvement in muscle function should be observed directly by the examiner. This test is most reliable when the patient has ptosis, ocular muscle weakness, or nasal speech, and results are positive in about 90% of such patients. Strength after Tensilon also may improve in motor neuron disease and in lesions of the oculomotor nerves.
When performing the Tensilon test, observe the degree of weakness in a select group of muscle functions for several minutes prior to administering Tensilon. The best Tensilon test results are often obtained in patients who have clear ptosis or ophthalmoparesis and a clear improvement can be observed following its administration. [31]
After an initial injection of 2 mg, the response in the selected muscles is monitored for 60 seconds. Subsequent injections of 2 mg are given at 60 second intervals for a total dose of 10 mg. If definite improvement is seen within 60 seconds after any dose, no further injections are necessary. Total dose in children is 0.15 mg/kg. The optimal dose of Tensilon varies among patients and cannot be determined in advance.
Bradycardia is a possible adverse effect, particularly in elderly patients, and ECG monitoring is advisable in this patient population. Atropine sulfate should be available at the bedside when performing the Tensilon test. Supportive respiratory measures should also be readily available when administering Tensilon because some patients are rather sensitive to even a small dose. Worsening weakness after administration of these doses of Tensilon (a paradoxical response) is also indicative of impaired neuromuscular transmission.
In the past, many have advocated also giving a placebo in a single-blinded fashion (ie, the patient does not know which injection is the Tensilon and which is the placebo). In cases in which either malingering or a conversion disorder is suspected, a placebo would be helpful in making this determination. In this situation, the Tensilon test may expose the patient to unnecessary risk. Usually, this has little use, and it should not be necessary with a well-defined endpoint, such as relief of ptosis.
Patients who do not respond to Tensilon may respond to intramuscular neostigmine (Prostigmin), which has a longer duration of action. This is particularly useful in infants and children, whose response to Tensilon may be too brief for adequate observation. Neostigmine inhibits the destruction of acetylcholine by acetylcholinesterase, which facilitates transmission of impulses across the myoneural junction.
In some, a therapeutic trial of oral pyridostigmine (Mestinon, Timespan, Regonol) for several days may produce improvement that cannot be appreciated after a single dose of Tensilon or neostigmine. [32]
Many patients with muscle-specific tyrosine kinase antibody (MuSK-Ab)-positive MG, a variant, do not improve with Tensilon or pyridostigmine.
The Tensilon test result is sometimes positive in botulism poisoning, amyotrophic lateral sclerosis (ALS), and in some cases of myopathy. Generally, the history and other aspects of the context of the presentation aide the examiner in determining the cause of a positive Tensilon test result. [33]
Immunologic testing
The following 3 antibody tests are now available to aid in the diagnosis of MG:
Anti-acetylcholine receptor antibody (anti-AChR Ab)
Muscle-specific tyrosine kinase antibody (MuSK Ab)
LRP4 antibodies
The anti-AChR antibody test is the mainstay of immunologic testing. The test exhibits a high sensitivity and an extremely high specificity. The sensitivity varies with the clinical presentation of the MG. For patients with generalized MG, the sensitivity is roughly 85-90%. For ocular MG, sensitivity is 50-60%. The specificity is very high, although, interestingly, false-positive results are not uncommon in Lambert-Eaton myasthenic syndrome (LEMS). False-positive results are also seen in thymoma without MG and lung cancer.
Some of the patients with MG who previously tested negative for anti-AChR antibodies, later test positive for antibodies to muscle-specific tyrosine kinase (MuSK). The percentages vary from series to series, but roughly 38-50% of patients who test negative for the anti-AChR Ab will have a positive anti-MuSK test result. [34, 35, 36, 37] Patients with positive anti-MuSK MG are predominantly women. [38, 39] The distribution of muscle involvement may be different than that seen in the more common AChR antibody-positive MG. One group of authors has found that patients testing positive for anti-MuSK display significant oculobulbar involvement less frequently than typical anti-AChR-positive patients and more frequently show neck, shoulder, and respiratory muscle involvement. In addition, they found no purely ocular cases. [38]
A third set of antibodies, antibodies against low-density lipoprotein receptor-related protein (LRP4), an agrin receptor may be positive in cases that do not test positive for anti-AChR Ab or anti-MuSK Ab. This is a smaller porportion compared to the other two antibodies. [40, 41]
Another disease entity, called seronegative MG, was previously used in patients who did not possess the anti-AChR Ab. However, this term is now more precisely defined, as its name implies, as patients who are seronegative for all three: anti-AChR Ab, anti-MuSK Ab, and LRP4 antibodies. This represents about 6-12% of the population with MG. Clinically, these patients tend to have mainly ocular manifestations and tend to respond well to the various therapies used in the treatment of MG. [35, 42]
Repetitive Nerve Stimulation
Electrodiagnostic studies are used to demonstrate abnormal neuromuscular transmission and to exclude other diseases of the motor unit that mimic or contribute to the clinical findings. Such studies may also be useful in measuring the severity of involvement and demonstrating changes as the disease evolves or improves.
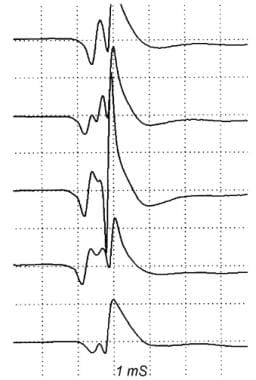
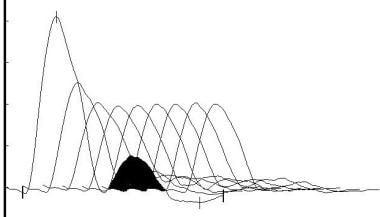
The most commonly used electrodiagnostic test of neuromuscular transmission involves repetitive stimulation of a motor nerve (RNS) while recording compound muscle action potentials (CMAP) from a muscle innervated by that nerve. This is essentially the Jolly test originated by Dr Friedrich Jolly, as mentioned in the Introduction. In his recordings, he quantified the force produced by the muscle rather than the CMAP (however, the force generally correlates with the CMAP). The result is abnormal if progressively fewer muscle fibers respond to nerve stimulation during a train of stimuli, producing a "decrementing" pattern in the CMAP.
A typical decrementing response to repetitive nerve stimulation in myasthenia gravis. The amplitude of the initial response is normal, and the decrement is maximal in the fourth response. Thereafter, the responses increase somewhat, giving a U-shaped envelope to the train of responses.
Although the technique is straightforward, it is not without technical pitfalls for the unwary. Most common errors result from movement of the recording electrode, variations in the stimulus intensity, and inadequate warming of the muscle.
Recording technique
Surface recording electrodes are placed so that an initial sharp negative (upward-going) deflection of the CMAP is produced by supramaximal nerve stimulation, indicating that the active electrode is over the motor point of the muscle. [43] The reference-recording electrode should be over a distal point where electrical activity from the muscle is minimal. Intramuscular needle electrodes should not be used to record the responses, since they cannot be kept at a constant position within the muscle during nerve stimulation. Appropriate joints should be immobilized to minimize movement artifact.
Muscle temperature
Decremental response in MG is lessened when the muscle is cool. When the abnormality is mild, no decrement may be seen unless the muscle is warmed. Hand or foot muscles should be warmed to a surface temperature of at least 34°C to obtain maximum diagnostic sensitivity. Cooling is not as great a factor in proximal muscles, which need not be warmed before testing.
Stimulation technique
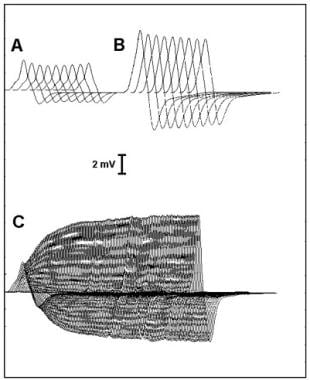
Stimulation of the motor nerve usually is performed with surface electrodes. Stimulation also can be performed with near-nerve needle electrodes, in which case, shorter-duration and lower-intensity stimulation pulses may be used, which are less painful when the needles are placed optimally. To ensure maximal responses throughout the testing session, the stimulus intensity should be 10-25% greater than required to activate all muscle fibers. A decremental response in MG is best demonstrated at stimulation rates of 3-5 Hz. Some laboratories also use 2 Hz. Decrement increases with the stimulation rate, up to 10 Hz. At rates greater than this, the CMAP size may increase during stimulation (ie, potentiation), and artifacts due to movement are common. Amplitude increments up to 50% have been reported in normal muscle with stimulation at rates of 10-50 Hz.
Potentiation may result from an increase in the number of activated muscle fibers (ie, facilitation) or from an increase in the amplitude or summation of action potentials of the individual muscle fibers (ie, pseudofacilitation).
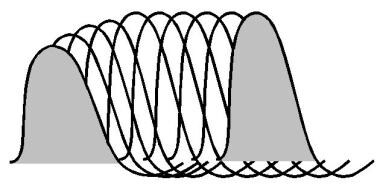 A diagram of pseudofacilitation in the muscle responses seen during high-frequency repetitive nerve stimulation. Amplitude of the responses increases due to shortening of the waveforms, but the area under the waveforms remains relatively constant.
A diagram of pseudofacilitation in the muscle responses seen during high-frequency repetitive nerve stimulation. Amplitude of the responses increases due to shortening of the waveforms, but the area under the waveforms remains relatively constant.
Facilitation results when 2 or more nerve action potentials occur within a short time, producing a transient increase in the amount of ACh released from the motor nerve. Each nerve depolarization releases calcium into the periterminal space, which increases the local concentration of calcium for a short period of time. If the nerve depolarizes during this period, the increased calcium increases the number of ACh quanta released. In conditions of impaired neuromuscular transmission, this greater ACh release may improve synaptic transmission briefly, producing facilitation.
Pseudofacilitation has been attributed to increased synchronization of muscle fiber action potential propagation velocities or to hyperpolarization of the muscle fiber membrane from increased sodium-potassium pumping. In normal muscle, pseudofacilitation may increase the CMAP amplitude 50% during stimulation at rates up to 50 Hz. This can mask a decrementing response or may be mistaken for true facilitation.
Measurement technique
The size of the CMAP may be assessed by measuring either the amplitude or the area of the negative peak of the CMAP. The CMAP should be observed at a fast display sweep speed (50-100 msec sweep duration) during repetitive stimulation to detect technical artifacts. Movement of the stimulating electrode or the muscle may produce irregular patterns of change in CMAP size during trains of stimuli (these do not correspond to patterns expected in disease). In MG, the typical pattern is a progressive decrement of the second through the fourth or fifth response, with some return toward the initial size during the subsequent responses, a so-called U-shaped pattern (see image below). This pattern also is seen in Lambert-Eaton myasthenic syndrome (LEMS), motor neuron disease, and rarely in peripheral nerve disease.
A typical decrementing response to repetitive nerve stimulation in myasthenia gravis. The amplitude of the initial response is normal, and the decrement is maximal in the fourth response. Thereafter, the responses increase somewhat, giving a U-shaped envelope to the train of responses.
Changes in CMAP size are quantified by calculating the percentage change in amplitude (or area) between the first and a later response, usually the fourth or fifth response in a train.
Stimulation at rates of 3-5 Hz may produce a decrement up to 8% in normal muscles. A decrement greater than 10% usually is abnormal. Even a decrement of 5% may be considered abnormal if artifacts demonstrably are not responsible for the change. The following observations help distinguish true decrements from artifactual changes in CMAP size:
-
Reproducibility: Decrement should be the same when stimulation is repeated after a period of rest.
-
Envelope shape: Changes during a train of stimuli should conform to a pattern seen in disease, without sudden or irregular variations between consecutive responses.
-
Activation cycle: Changes induced by activation (see Activation below) should conform to an acceptable pattern.
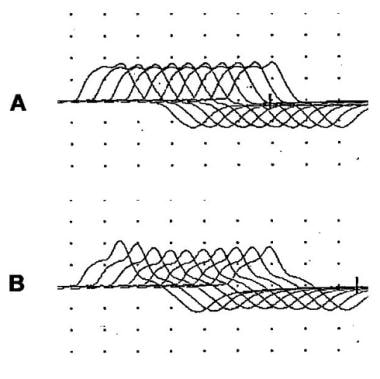
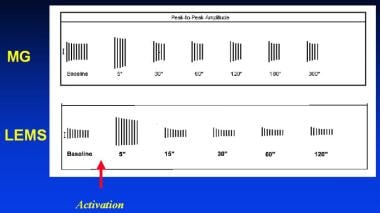 Typical activation cycles seen during repetitive nerve stimulation. The vertical lines represent the amplitude of muscle responses during 3-Hz repetitive nerve stimulation before and at indicated intervals after activation of the muscle by brief maximum voluntary contraction (arrow). In myasthenia gravis (MG), a decrementing response is observed initially, which partially repairs after activation and is later exaggerated. In Lambert-Eaton myasthenic syndrome (LEMS), the responses are small. A decrementing response occurs, and activation is followed by a marked and brief increase in response amplitude.
Typical activation cycles seen during repetitive nerve stimulation. The vertical lines represent the amplitude of muscle responses during 3-Hz repetitive nerve stimulation before and at indicated intervals after activation of the muscle by brief maximum voluntary contraction (arrow). In myasthenia gravis (MG), a decrementing response is observed initially, which partially repairs after activation and is later exaggerated. In Lambert-Eaton myasthenic syndrome (LEMS), the responses are small. A decrementing response occurs, and activation is followed by a marked and brief increase in response amplitude.
-
Response to Tensilon: Administration of intravenous Tensilon usually reduces or obliterates a decrement that is due to abnormal neuromuscular transmission. In some cases, a paradoxical response to edrophonium may be observed in which a decrement worsens or appears only after administration of Tensilon. Either response indicates that neuromuscular transmission is abnormal.
-
Activation: A period of nerve activation is followed by 2 consecutive phases. During the first, facilitation, each nerve impulse releases more ACh than before activation. During the second, postactivation exhaustion, less ACh is released by each nerve impulse. The exhaustion is maximum 2-5 minutes after the end of activation. The nerve may be activated by high-frequency nerve stimulation (20-50 Hz) or by having the patient contract the muscle maximally for 10-30 seconds; the latter is less painful.
Depending on what disease one is expecting, the length of the activation period may be varied. Generally, 10 seconds is enough to demonstrate facilitation in LEMS. For MG, 10 seconds may also be enough to get the facilitation but not enough to show a definitive period of exhaustion, which is also very helpful in the diagnosis. Thus, at least 30 seconds of voluntary activation is frequently used. Even up to 2 minutes may be required to produce exhaustion in MG. Trains of low-frequency stimuli are given within 10 seconds after the end of activation to demonstrate facilitation and at intervals for up to 10 minutes thereafter to demonstrate postactivation exhaustion.
Muscle selection
RNS is more likely to be abnormal in proximal or facial muscles, owing to the fact that they are more involved in the disease process. To obtain the maximum diagnostic yield, examination of several muscles, including those that are involved clinically, may be necessary.
-
Hand muscles are the most convenient to test. Recordings can be made from the hypothenar or first dorsal interosseous muscles while stimulating the ulnar nerve at the wrist or from the thenar muscles while stimulating the median nerve. Stimulating the musculocutaneous nerve in the axilla tests the biceps. This nerve can be stimulated with surface electrodes pressed firmly against the posterior border of the short head of the biceps within 1 inch of the inferior edge of the axillary fold. Such stimulation may be uncomfortable, especially if prolonged or high-frequency trains are used. A needle-stimulating electrode inserted near the nerve reduces discomfort and is less likely to be dislodged during stimulation and activation.
-
To test the deltoid muscle, the active recording electrode is placed on the belly of the muscle and the reference on the acromion. Stimulation is performed at the Erb point with a surface electrode pressed firmly behind the clavicle. This stimulation is uncomfortable, and stimulating other muscles cannot be avoided.
-
The trapezius is perhaps the easiest shoulder muscle to test (see image below). The patient is examined while seated on a chair with the arm hanging straight down. The active recording electrode is placed on the trapezius at the angle between the neck and the shoulder, and the reference electrode is placed on the acromion. The spinal accessory nerve is stimulated at the posterior border of the sternocleidomastoid muscle behind the ear. The nerve is superficial at this point and can be maximally stimulated with low-intensity pulses, which minimize discomfort and avoid stimulating other muscles.
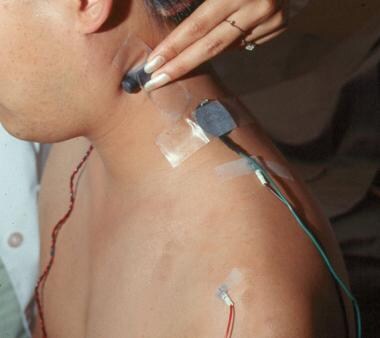 Electrode positions for performing repetitive nerve stimulation of the trapezius muscle. The active recording electrode (black) is placed over the belly of the muscle and the reference-recording electrode (red) distally over the shoulder. The spinal accessory nerve is stimulated as it crosses the sternocleidomastoid muscle. The green wire is the ground.
Electrode positions for performing repetitive nerve stimulation of the trapezius muscle. The active recording electrode (black) is placed over the belly of the muscle and the reference-recording electrode (red) distally over the shoulder. The spinal accessory nerve is stimulated as it crosses the sternocleidomastoid muscle. The green wire is the ground.
-
Facial muscles are tested with the active electrode over the nasalis and the reference electrode lateral to the eye. Alternatively, the inferior orbicularis can be studied, with the reference over the nose. The facial nerve is stimulated below the ear. However, movement artifact frequently limits the interpretation of responses from facial muscles, and many patients may not tolerate the stimulation.

Methods to enhance or provoke a decrementing response
If no decrement is seen before or after activation, mild abnormalities in MG may be revealed during the exhaustion period that follows prolonged nerve stimulation at slow rates (3 Hz for 4-5 min). Producing ischemia in the tested muscle during the period of prolonged stimulation and subsequent exhaustion may enhance the sensitivity of this technique further. To perform this, a blood pressure cuff is placed around the upper arm and inflated to above systolic pressure, and a distal nerve is stimulated. In patients with MG who have normal RNS studies in the hand muscles, this enhancing technique has been shown to be slightly more sensitive than repetitive stimulation of the trapezius; however, it is only 60% as sensitive as single-fiber electromyography (EMG) of the hand muscle. [44]
Electromyography
Conventional needle electromyography
Study of the motor unit action potentials (MUAPs) with the standard concentric or monopolar EMG needles often provides considerable information about transmission at the neuromuscular junction. The MUAP demonstrates variability in the shape or amplitude.
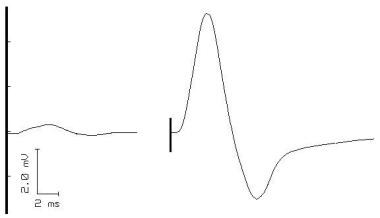
 Action potentials from a single motor unit in a patient with Lambert-Eaton myasthenic syndrome. The waveforms vary from discharge to discharge, indicating abnormal neuromuscular transmission.
Action potentials from a single motor unit in a patient with Lambert-Eaton myasthenic syndrome. The waveforms vary from discharge to discharge, indicating abnormal neuromuscular transmission.
This occurs, because with a neuromuscular junction problem, the muscle fibers that are part of the motor unit do not always fire in the same sequence. In addition, some of them fail to fire. The fact that the sequence varies is reflected in the idea of "jitter," which is discussed in the next section (single-fiber electromyography). The fact that sometimes some of them fail to fire at all is actually the same thing as the "blocking" discussed in single-fiber electromyography. Unstable MUAPs also are observed in denervating disease, especially motor neuron disease; thus; MUAPs are not specific for MG. When seen without evidence of neuronal disease, unstable MUAPs should prompt an assessment for MG or other disease of neuromuscular transmission. [45]
Single-fiber electromyography
Single-fiber EMG (SFEMG) is the most sensitive electrodiagnostic test of neuromuscular transmission. SFEMG demonstrates increased jitter in a limb or face muscle in almost all patients with MG. Because of its marked sensitivity, SFEMG also demonstrates abnormal jitter in other nerve and muscle diseases; thus, the results must be interpreted in conjunction with the results of more conventional electrodiagnostic studies.
The details of this technique are well described in Single-Fiber EMG. However, some basic principles of the single-fiber method are described below.
The single-fiber technique was developed by Stalberg and Ekstedt in order to study muscle fatigue. It is the most sensitive test of dysfunction at the neuromuscular junction. Ordinarily, it requires a special needle electrode. The needle is thin, 25 μm in diameter, but no thinner than most monopolar needles or the smaller concentric needles. Its configuration is quite unusual. Although it has a pointed bevel at the very tip, the opening for the recording surface is located on the side, a bit proximal (3-5 mm) to the tip. The recording surface is very small, and the volume of tissue from which it effectively records is tiny, only about 300 μm3 compared to 1 cm3 for most standard concentric needles.
Another factor that limits the range from which significant potentials can be recorded is the filter setting. Single-fiber EMG typically uses a 500-Hz high-pass filter. This filter attenuates the lower frequency distant volume-conducted potentials. [46, 47]
Because of the small size of the area sampled, a single-fiber needle is able to pick up and discriminate potentials from single nearby muscle fibers, as opposed to the composite motor unit potential (MUP) (also called motor unit action potential [MUAP]), which is picked up by the standard monopolar or concentric EMG needle.
The examiner asks the patient to contract a muscle slightly with the single-fiber needle in place. Under those circumstances, the needle usually records a single MUP at a time. However, occasionally, it picks up two MUPs that fire at slightly different times. Because the recording volume of the needle is so small, it is a good bet that the two MUPs are coming from two individual muscle fibers that are very close together and which are part of the same motor unit. As the muscle continues to contract, one might expect that if muscle fiber Ma contracted T microseconds before muscle fiber Mb, then each time Ma contracts Mb will contract exactly T microseconds later.
Of course, biological processes always have some variation, but if one tries to keep all conditions the same, the differences should be small. Indeed, for normal neuromuscular junctions they are small, usually in the range of 10-50 microseconds (ms). However, when a problem occurs with the function of the neuromuscular junction, then the variation is greater. This variation is called the jitter. The test result is positive when the jitter significantly exceeds the norm. It is highly sensitive for MG (at least 90% and probably 95%) but not very specific.
A second type of abnormality that may be observed is called blocking. Assuming that one has found a pair of fibers that tend to fire one after the other, blocking is the failure of 1 of the 2 to fire at all. One could look at it as the extreme case of exaggerated jitter (ie, the interval goes to infinity). If blocking occurs, then actual weakness of the muscle should be present because this means that some of the fibers that should be consistently firing and contracting are not always contracting. Muscles with a significant amount of blocking should also show a decrement when tested with repetitive stimulation. However, muscles with only an increased jitter will generally not show significant weakness, and they are the ones that are generally negative (ie, normal) on repetitive stimulation but abnormal on single-fiber studies. The excessive jitter and the blocking contribute to an unstable appearance of the MUAPs in MG and other diseases of neuromuscular transmission. [48, 49, 50, 51]
Stimulated single-fiber EMG
Some patients are unable to voluntarily contract the muscle in order to allow SFEMG to be performed. In those cases, often the examiner can produce the contraction by electrically stimulating a branch of the nerve innervating the muscle. See Single-Fiber EMG for more detail.
RNS Versus Single-Fiber EMG
RNS demonstrates an abnormal decrement in a hand or shoulder muscle in 75% of patients with generalized MG and in only 50% of patients with ocular MG. Testing a facial muscle increases the yield somewhat, especially in patients with weakness that predominates in these muscles.
SFEMG demonstrates abnormal jitter in at least 1 muscle in 99% of patients with generalized MG and in 97% of those with ocular MG. Increased jitter and blocking frequently are found in muscles in which no decrement is seen on RNS, but the converse is not observed.
SFEMG is most valuable clinically in the patient with suspected MG in whom other tests of neuromuscular transmission and AChR antibody measurements are normal. When abnormal neuromuscular transmission has been demonstrated by RNS, the finding of abnormal jitter does not add to the diagnosis, although baseline jitter values may be useful for comparison with subsequent studies.
In patients with MUSK-positive MG, RNS and SFEMG studies are frequently normal in commonly tested muscles, and examining the most severely involved muscles to demonstrate a decremental response or abnormal jitter may be necessary.
Lambert-Eaton Myasthenic Syndrome
LEMS, is described in detail in Lambert-Eaton Myasthenic Syndrome. However, key points highlighting neuromuscular transmission are discussed here.
LEMS is caused by autoantibodies to the voltage-gated calcium channels (VGCC) on the presynaptic side of the neuromuscular junction. Frequently, it is a paraneoplastic disease, with the most common being small-cell carcinoma of the lung (SCLC). Tests for these antibodies are commonly available.
Bulbar weakness is usually absent in those with LEMS. Appendicular weakness is commonly encountered in the proximal musculature of the legs. Typically, it is not uncommon for patients with LEMS to report an increase in strength with repeated exercise. With continued exercise, however, weakness eventually ensues. In about one half of patients, an increase in strength with exercise does not occur. [52, 53]
In those afflicted with LEMS, repetitive nerve stimulation discloses small CMAP amplitudes (often less than 10% of normal) and a decremental pattern is observed with low-frequency stimulation. See the image below.
 Repetitive nerve stimulation studies in Lambert-Eaton myasthenic syndrome. The initial response amplitude is low, and a decrementing response to low-frequency stimulation is noted (A). Immediately after activation, the amplitude is increased (B). During high-frequency stimulation (C), the initial decrement is followed by an increase in amplitude to more than twice the initial value.
Repetitive nerve stimulation studies in Lambert-Eaton myasthenic syndrome. The initial response amplitude is low, and a decrementing response to low-frequency stimulation is noted (A). Immediately after activation, the amplitude is increased (B). During high-frequency stimulation (C), the initial decrement is followed by an increase in amplitude to more than twice the initial value.
The most characteristic electrodiagnostic feature is facilitation, which is an increase in the size of the CMAP after activation by maximum voluntary contraction or during stimulation at rates of at least 20 Hz. The degree of facilitation varies considerably among patients with LEMS. Findings may be masked partially by low muscle temperature; thus, tested muscles should be warmed for best results. [54] Facilitation greater than 100% (doubling of the CMAP from resting values) is most commonly observed in hand muscles. Not all muscles show this pattern; in some patients with LEMS, facilitation may be less than 100% in all muscles tested.
Characteristic findings are demonstrated in most patients with LEMS by the following protocol:
Measure the size (amplitude or area) of the CMAP elicited from the rested muscle by supramaximal nerve stimulation. Warming of the muscle to at least 34°C surface temperature and resting for several minutes before testing is essential.
Have the patient contract the tested muscle maximally for 10 seconds and then relax completely. Deliver a supramaximal nerve stimulus as soon as relaxation is complete. This stimulus should be delivered as soon as possible after the end of muscle activation, preferably within 5 seconds. Compare the size (amplitude or area) of the CMAP with that obtained in the rested state (see image below). The most consistent results will be obtained if this sequence is repeated and the average of at least 3 responses is used to assess the degree of facilitation.
After several minutes of relaxation, deliver a train of low-frequency (2-5 Hz) stimuli to the nerve and measure the percentage decrement.
 Muscle responses to nerve stimulation in a patient with Lambert-Eaton myasthenic syndrome. The initial response (left) is very small. After brief maximum voluntary contraction, the amplitude (right) increases markedly.
Muscle responses to nerve stimulation in a patient with Lambert-Eaton myasthenic syndrome. The initial response (left) is very small. After brief maximum voluntary contraction, the amplitude (right) increases markedly.

Perform these measurements in at least 1 hand and 1 foot muscle. If facilitation less than 100% is seen in a hand muscle, another hand muscle should be tested. Nerve stimulation at 20 Hz may be used instead of voluntary contraction to activate the muscle, although the authors find the voluntary contraction technique to be more sensitive and less painful. The size of the maximum CMAP after stimulation for 5-7 seconds is compared with that of the initial response (see image below). Measuring the area of these CMAPs is difficult, so the amplitudes usually are measured instead.
 Typical activation cycles seen during repetitive nerve stimulation. The vertical lines represent the amplitude of muscle responses during 3-Hz repetitive nerve stimulation before and at indicated intervals after activation of the muscle by brief maximum voluntary contraction (arrow). In myasthenia gravis (MG), a decrementing response is observed initially, which partially repairs after activation and is later exaggerated. In Lambert-Eaton myasthenic syndrome (LEMS), the responses are small. A decrementing response occurs, and activation is followed by a marked and brief increase in response amplitude.
Typical activation cycles seen during repetitive nerve stimulation. The vertical lines represent the amplitude of muscle responses during 3-Hz repetitive nerve stimulation before and at indicated intervals after activation of the muscle by brief maximum voluntary contraction (arrow). In myasthenia gravis (MG), a decrementing response is observed initially, which partially repairs after activation and is later exaggerated. In Lambert-Eaton myasthenic syndrome (LEMS), the responses are small. A decrementing response occurs, and activation is followed by a marked and brief increase in response amplitude.
In patients with LEMS, MUAPs are unstable and vary in shape during voluntary activation. The jitter measured by SFEMG is increased markedly, frequently out of proportion to the severity of weakness; impulse blocking is frequent. In many endplates, jitter and blocking decrease as firing rate increases. This pattern is not pathognomonic for LEMS and may be observed in some endplates in patients with MG. Thus, improvement of jitter and blocking at higher activation rates (unless dramatic) does not indicate a presynaptic abnormality. [55]
Overlap syndrome
The seemingly opposite nature of some of the MG findings versus the LEMS findings are not invariably found. Either one of these diseases can somehow imitate the other in what is sometimes called an overlap syndrome. Thus, no single clinical or electromyographic feature distinguishes MG from LEMS in all patients. Some patients with clinically typical MG have facilitation greater than 50% or even up to 100% in some muscles at high stimulation rates. Conversely, patients with LEMS may have EMG features more characteristic of MG at some point during their illness.
In other patients, because of mixed clinical and EMG features, distinguishing between these 2 diseases may not be possible. Patients with clinical features typical for MG (including elevated AChR antibodies) have been observed to have an RNS pattern characteristic of LEMS at one time and a pattern typical for MG in the same muscle at another time. In patients with such mixed features, the clinical characteristics and the presence of AChR antibodies or lung cancer define the most likely diagnosis.
Congenital (Genetic) Myasthenic Syndromes
Several forms of myasthenia are due to genetic defects and are not associated with any immunologic abnormality. Some of these conditions present at birth, whereas other, milder forms of the disease often do not come to clinical attention until adulthood. These diseases do display characteristic electrodiagnostic findings.
Congenital myasthenia
Congenital myasthenia is a clinical term used to describe patients with one of several genetic neuromuscular defects who usually have ophthalmoparesis and ptosis at birth or shortly thereafter. Electrodiagnostic findings are similar to those of acquired MG. RNS studies demonstrate a decremental response, which is corrected by edrophonium, and SFEMG shows increased jitter. Intracellular recordings of muscle biopsy samples must be obtained to determine the physiologic abnormality and the specific diagnosis in these patients. In some of these diseases, the specific gene is now known, and the potential for diagnosis via genetic testing exists.
Classification of these disorders is based on the specific defect found at the neuromuscular junction; that is, whether or not presynaptic, synaptic, or the postsynaptic machinery is afflicted. The presynaptic variety is the rarest of the 3, affecting an estimated 7-8% of patients. [56]
This is an area of active research, and new mutations continue to be discovered. Listed below are some of the major variants together with their Online Mendelian Inheritance in Man (OMIM) number. OMIM is a comprehensive human gene catalog edited by Dr. Victor A. McKusick and his colleagues and is available on the Web as a result of the efforts of the National Center for Biotechnology Information (NCBI). [57]
Congenital myasthenic syndromes coupled to presynaptic defects
End-plate ChAT deficiency (congenital myasthenic syndrome associated with episodic apnea)
Congenital myasthenic syndrome associated with episodic apnea is also called familial infantile myasthenia (OMIM #254210). This form of congenital myasthenia has unique clinical and electrophysiological features. Physiological studies in familial infantile myasthenia have demonstrated findings consistent with abnormalities of acetylcholine synthesis, resynthesis, or mobilization. The Mayo Clinic group has identified many specific mutations in the choline acetyl transferase (ChAT) gene on arm 10q that give rise to this autosomal recessive condition. [58] However, another mutation on chromosome 17 (near or actually in the gene for synaptobrevin-2) appears to produce the same phenotype. Clinically, these patients manifest episodes of bulbar weakness and respiratory distress, resulting in apnea. Often times, pyrexia or infection causes the apneic symptoms to manifest.
Weak muscles usually have a decremental response to RNS. Strong muscles may have a decremental response only after sustained voluntary muscle activity or prolonged nerve stimulation (see image below), performed as follows: Stimulate the nerve at 3-5 Hz for 5 minutes, then deliver brief trains of 3- to 5-Hz stimuli. These trains demonstrate a decrement that clears over a 20- to 30-minute period and can be corrected by Tensilon. [59]
 Muscle responses to 3-Hz nerve stimulation in a hand muscle of a patient with familial infantile myasthenia. The initial response (A) is normal, but a decrementing response occurs after prolonged 3-Hz stimulation (B).
Muscle responses to 3-Hz nerve stimulation in a hand muscle of a patient with familial infantile myasthenia. The initial response (A) is normal, but a decrementing response occurs after prolonged 3-Hz stimulation (B).
Progressive neuromuscular blockade also can be demonstrated in these patients with SFEMG, which shows increasing jitter during prolonged nerve stimulation.
Treatment consists of pyridostigmine or 3,4-diamniopyridine (3-4,DAP).
Congenital myasthenic syndrome with a paucity of synaptic vesicles
In this disease, whose detailed pathophysiology and genetic mechanism have yet to be fully described, there are decreased total amounts of acetylcholine synaptic vesicles with resultant decreased release of acetylcholine quanta. The probability of quantal release remains unaffected. [58] Ophthalmoplegia, ptosis, and hypotonia are common presenting signs.
"Lambert-Eaton-like congenital myasthenic syndrome"
There is marked facilitation of the cMAP with repetitive stimulation, hence the name of this disease. However, there is no identifiable defect in the genes that encode for calcium channels. There is a decreased probability of quantal release, but the ultrastructural mechanisms, as evidenced by endplate study, do not reveal any defects. The genetic mechanisms underlying this disease remain occult at present. [58]
Congenital myasthenic syndromes coupled to synaptic defects
Endplate acetylcholinesterase deficiency
In endplate acetylcholinesterase deficiency (OMIM #603034), there is weakness of facial, oropharyngeal, neck, and limb muscles seen in the neonatal period (or shortly thereafter). Patients show generalized weakness, which is exacerbated by exertion. Frequently, the pupillary light reflex is slowed (or even absent), and ophthalmoparesis may be present. Scoliosis or lordosis may also be present. The severity of the different symptoms varies considerably from patient to patient.
The disease is caused by an autosomal recessive gene (COLQ; 603033) that codes for the collagen-like tail of endplate acetylcholinesterase. Histochemical stains show that acetylcholinesterase is absent from the nerve terminals, which are reduced in size and exhibit a reduced postsynaptic membrane density and degeneration of the postsynaptic folds. An end-plate myopathy is encountered as well, with a proclivity toward type II fiber atrophy. Antiacetylcholinesterase medications such as physostigmine have no beneficial effect, as one would expect; sometimes, they have a detrimental effect (possibly because a small amount of residual acetylcholinesterase activity exists, which is blocked). Albuterol has been used with some success in this disease.
RNS produces a decrement on 2-Hz stimulation in all muscles. Single-nerve stimulation produces repetitive CMAPs 6-10 milliseconds after the initial response, which fade quickly during repetitive stimulation, even at rates as low as 0.2 Hz. This is similar to findings in slow channel syndrome.
 Responses to 3-Hz nerve stimulation in a hand muscle in a patient with slow channel syndrome. A decrementing pattern is noted, and a repetitive discharge (black) is seen following the response to the initial stimulus.
Responses to 3-Hz nerve stimulation in a hand muscle in a patient with slow channel syndrome. A decrementing pattern is noted, and a repetitive discharge (black) is seen following the response to the initial stimulus.
In routine electrodiagnostic studies, CMAP after-discharges are often encountered.
Congenital myasthenic syndrome associated with beta-2-laminin chain deficiency
In this disorder, there is a defect of the LAMB2 gene, which encodes for beta-2 laminin protein, which is a component of the neuromuscular junction. This protein is also found in the eye and the kidney. Electrophysiologically, the disease manifests decreased endplate potential quantal release and miniature endplate potential frequency. Electron microscopy discloses widened synaptic spaces and simplified junctional folds. [56, 58] Patients with this defect have Pierson syndrome (OMIM # 609049), which results in miosis due to atrophy of the dilatator pupillae muscle as well as nephrotic syndrome in the neonatal period. One patient described in the literature had a heteroallelic missense and frameshift mutation in LAMB2 and possessed not only the ocular and renal manifestations, but severe proximal limb, ocular muscle, and respiratory muscle weakness as well. Treatment is with ephedrine. [58]
Congenital myasthenic syndromes coupled to postsynaptic defects
Primary kinetic abnormalities of the acetylcholine receptor
Two varieties exist: slow-channel and fast-channel syndromes.
Slow channel syndrome
In slow channel syndrome (OMIM #601462), the underlying physiological cause of the abnormalities is a prolonged open time of the AChR ion channel (ie, the channel closes too slowly, hence the name slow channel syndrome), resulting in calcium ion entry. This can be caused by a number of mutations. Typically, they are autosomal dominant mutations in the alpha, beta, delta, or epsilon subunits of the AChR. Rarely, the mutation is autosomal recessive (and in that case, it is usually an epsilon mutation). This syndrome may not produce weakness until the patient is an adult, which makes distinguishing it from acquired MG difficult. Onset can range from the first to sixth decade. As most of the cases are autosomal dominant, if a dominant family history is found in what otherwise looks like acquired MG, this extremely rare condition needs to be considered. [60, 61, 62, 63, 64]
Clinically, there is ptosis as well as finger extensor and cervical axial weakness.
RNS studies produce a decremental response. Characteristic repetitive discharges are seen after single-nerve stimulation in most, but not all, muscles. Electrodiagnostic findings are similar to those seen with cholinesterase inhibitor toxicity and congenital acetylcholinesterase deficiency.
Responses to 3-Hz nerve stimulation in a hand muscle in a patient with slow channel syndrome. A decrementing pattern is noted, and a repetitive discharge (black) is seen following the response to the initial stimulus.
Treatment consists of fluoxetine in high doses. Fluoxetine actually blocks the Ach-R channel. Quinidine and pyridostigmine have also been used with efficacy. [65]
Fast channel syndrome
The clinical manifestations of fast channel syndrome (OMIM #608930) are somewhat variable. Usually, patients have weakness of ocular muscles with mild-to-moderate weakness of musculature of the face, neck, trunk, limbs, and respiratory muscles. Like most other types of myasthenia, there is easy fatigability. During infancy, children frequently manifest with poor effort in crying and hypotonia.
The disease is usually autosomal recessive, but some autosomal dominant cases have been found. The problem is caused by mutation in certain subunits of the acetylcholine receptor such as alpha 1, epsilon, and delta. [66]
On electron microscopy, the morphology appears intact: the synapses look normal with normal-appearing junctional folds and a normal distribution of acetylcholine receptors. Some response to anti-acetylcholinesterase medications, such as pyridostigmine, occurs, but a better response occurs to 3,4-diaminopyridine (3,4-DAP).
The basic neurophysiology is intriguing. The kinetics of the openings and closings of the channel are roughly the opposite of what is seen in slow channel syndrome (ie, in fast channel syndrome, it opens too slowly and closes too fast). Fewer, shorter openings are available. Thus, there is overall a decreased number of openings and an even greater decrease in the channel's open time, leading to small miniature endplate potentials (MEPPs) and small miniature endplate currents (MEPCs). However, quantal content (amount of acetylcholine in each vesicle) is normal.
In clinical neurophysiologic studies, decremental responses are seen on repetitive stimulation (but not very many patients have been studied). The authors could not find published studies of single-fiber EMG in fast channel syndrome. From the physiology, it would be expected to be abnormal.
In the hereditary congenital myasthenia, mutations are found in all the known important parts of the neuromuscular junction: AChR (acetylcholine receptor) mutations; mutations in one of the associated membrane proteins (rapsyn); AChE (acetylcholinesterase) mutations; Chat (choline acetyl-transferase) mutations; and mutations in (or near) synaptobrevin, one of the proteins involved in vesicle fusion. [67, 68]
Acetylcholine receptor deficiency without kinetic abnormalities
The symptoms of this disease, namely ptosis, ophthalmoplegia, dysphagia, muscular atrophy, lordosis, and scoliosis, can manifest over a wide range of ages from childhood to adulthood. Inheritance is autosomal recessive in females afflicted with this disease. The CHRNE gene, which encodes for the epsilon subunit of the acetylcholine receptor, is most commonly found. [56, 58] Genetic testing is available.
Congenital myasthenic syndrome caused by defects in the acetylcholine receptor complex
There are a few post-synaptic congenital myasthenic syndromes that are due to defects in rapsyn, Dok-7, and MuSK proteins, all of which are involved acetylcholine receptor interactions with the postsynaptic cytoskeleton and result in an over-arching reduced function of the receptor itself. The 3 known protein defects manifest with different clinical presentations.
Rapsyn deficiency manifests in the neonatal period, often with skeletal deformities such as arthrogryposis, ptosis, bulbar weakness, and proximal-greater-than-distal muscle weakness.
In those with Dok-7 mutations, there is marked weakness of the proximal musculature, and it is also known as limb-girdle myasthenic syndrome. These patients present in the neonatal period with hypotonia, and there are often decreased fetal movements in utero. Electron microscopy in Dok-7 discloses endplate destruction with ongoing attempts at repair. Electrodiagnostic studies reveal decremental responses to repetitive stimulation.
In congenital myasthenic syndromes caused by MuSK defects, there is decreased acetylcholine receptor expression in the endplate and decreased endplate quantal content. There is a report in the literature of an individual who had myasthenic symptoms early in life, remitting in adolescence, only to experience recrudescence as a catamenial phenomenon. Genetic testing is available for confirmation for the aforementioned defects. [56, 58]
This is an area of ongoing research, with new mutations being discovered, and, as a result, reclassification of the individual diseases is an ongoing effort.
Botulism
Botulism is discussed in several other articles: emergency and terrorist aspects in CBRNE - Botulism, a pediatric perspective in Botulism, an infectious disease perspective in Botulism, and an ophthalmologic perspective in Botulism. Other articles on botulinum toxin and its uses include Botulinum Toxin (BOTOX®): Dystonia Treatment, BOTOX® Injections, Botulinum Toxin, and Botulinum Toxin: Overview.
First, the basic science facts about how botulinum toxin works are reviewed. Botulinum toxin is a presynaptic blocker. In order for the neuronal cell to release ACh into the synaptic cleft, the synaptic vesicles that contain the ACh must fuse with the membrane. This fusion requires a synaptic fusion complex that is composed of SNARE proteins. SNARE proteins are involved in the release of numerous neurotransmitters in many species from mammals through yeast, and they are part of a large superfamily of proteins. For ACh release at the neuromuscular junction, some of the key SNARE proteins are synaptobrevin, SNAP-25, and syntaxin I.
Botulinum toxin, which can bind to the nerve cell membrane and be taken up by endocytosis, cleaves some of these proteins. In particular, botulinum toxins A, C, and E cleave SNAP-25 (the commercially available Botox ® and Dysport ® are both type A). Types B, D, F, and G cleave synaptobrevin (Myobloc ® and Neurobloc® are type B). Type C also cleaves syntaxin. Any of the cleavages can block the formation of the complex, thus blocking ACh release and consequently blocking muscle contraction. [69]
The history and physical examination can provide many clues about whether botulism is a possibility. Remember the potential sources of the disease. As described by Taillac and Kim [70] (see CBRNE – Botulism), the categories include the following:
-
Food: Ask about recent food ingestion, especially canned goods or anything that could spoil.
-
Wounds: Visually check for wounds on physical examination.
-
Intestinal colonization in infants younger than 1 year: Laboratory studies of stool samples are indicated. Seek pediatric and pediatric GI consultation.
-
Rarely, intestinal colonization of older individuals with bowel problems such as colitis or in those who have had surgical procedures such as intestinal bypasses: Check for such problems through the patient’s history. Seek consultation as necessary.
-
Toxin injection, as either a consequence of excessive medical injections or deliberate poisoning: Ordinarily, it is very unlikely that anyone would develop significant life-threatening weakness from either Botox® or Dysport® given by competent licensed practitioners. However, rare cases have occurred in which unethical practitioners have used unapproved formulations of botulinum toxin that caused generalized neuromuscular weakness.
-
Inhalational botulism: This has occasionally occurred in laboratory workers. It could occur in deliberate individual poisoning (murder attempt) or during a terrorist attack. One should take enough of an occupational history to rule out occupational exposure. Physicians should use their own judgment regarding suspicions of poisoning and/or terrorism. Some index of suspicion is needed in any case of otherwise unexplained weakness.
With regard to physical (neurologic) findings, these are generally symmetric, and the "dozen D’s", as noted in the Taillac and Kim article (see CBRNE – Botulism), that summarize the neurologic involvement should be considered: "dry mouth, diplopia, dilated pupils, droopy eyes, droopy face, diminished gag reflex, dysphagia, dysarthria, dysphonia, difficulty lifting head, descending paralysis, and diaphragmatic paralysis." [70] The dilated pupils in particular and a poor pupillary contraction to light in general are not part of MG, though they are common findings in botulism poisoning. Dry mouth also is a feature of botulism that is not seen in MG (but it is seen in Lambert-Eaton myasthenic syndrome).
The Tensilon test may help direct the examiner toward botulism. Consider the physiology. Botulinum toxin blocks the release of ACh. Tensilon blocks the degradation of ACh. Thus, in theory, Tensilon should ameliorate the weakness of botulism, and it sometimes does. However, it cannot differentiate botulism from MG. The Tensilon test result is sometimes falsely positive in amyotrophic lateral sclerosis (ALS) and in some cases of myopathy. Generally, the history and other aspects of the context of the presentation will aide the examiner in determining the cause of a positive Tensilon test result. [71, 72]
With respect to blood tests, the criterion standard is presently a mouse toxicity assay. It is often necessary for the mouse to be observed for days. Thus, the neurophysiologic tests described below, which can be performed rapidly, are particularly valuable for investigation of poisoning. An enzyme-linked immunosorbent assay (ELISA) test has become available for rapid screening, though it is not as sensitive or specific as the mouse bioassay. More recently, a procedure using a combination of endopeptidase cleavage followed by mass spectroscopy (Endopep-MS) has been developed; [73] this test may ultimately become the new criterion standard. However, neurophysiologic testing will remain very important because it can be performed immediately in the office or at the bedside.
Electrophysiologic findings in botulism tend to evolve with time and may not be present early in the disease. EMG findings typical of botulism include the following:
-
Reduced CMAP amplitude in at least 2 muscles
-
At least 20% facilitation of CMAP amplitude during tetanic stimulation
-
Persistence of facilitation for at least 2 minutes after activation
-
No postactivation exhaustion
Not all patients with botulism will have the first finding. [74] If none are found, the diagnosis of botulism is unlikely. If all 4 are present, only hypermagnesemia is in the differential diagnosis (and that can be quickly proven or dispelled by blood Mg levels).
Generally, caveats exist with most checklists of diagnostic findings, and this is true of botulism. As has been pointed out by Maselli [75] and others, the findings in botulism can vary from case to case. CMAP amplitudes usually are reduced, but the reduction is not so striking as is seen in LEMS for example. The reduction could easily be in the range seen in patients with axonal neuropathies. Facilitation with tetanic stimulation is not always seen. If it is not seen, then one can not look for persistence of facilitation or exhaustion after the facilitation.
Other features that may be observed include a decrement to slow rate stimulation, fibrillations and/or positive sharp waves, and motor unit action potentials, which are short, small, and polyphasic. The motor units may also recruit early (ie, at relatively low firing rates and degrees of force) as is often seen in a myopathy. Many reasons for variation in diagnostic findings can exist. For example, Cornblath's group found a very high percentage of incrementing responses to high-frequency repetitive stimulation. [76] However, their cases were in infants who mostly had botulinum B toxicity. Studies of adults, who more commonly have botulinum A toxicity, have shown lower percentages of incrementation to high-frequency stimulation.
SFEMG demonstrates markedly increased jitter and blocking and has been abnormal in all reported cases of food-borne or wound botulism. Jitter and blocking may decrease as the firing rate increases, but this is not a consistent finding.
Anti-Acetylcholinesterase Toxicity
The most common cause of poisoning by anti-acetylcholinesterases is accidental poisoning via inadvertent pesticide ingestion. In addition to such pesticides as malathion and parathion, chemists have created nerve agents such as sarin and soman, which are much more potent and are designed to be used for military purposes. They also have fallen into the hands of terrorists.
These agents work by stopping endogenous acetylcholinesterases from inactivating acetylcholine at the synapse. This causes excess acetylcholine to accumulate. So much acetylcholine accumulates that the muscles are in a constant state of depolarization causing depolarization block. Most such agents also cause central and GI effects with excess acetylcholine accumulation at muscarinic receptors. In fact, the muscarinic effects tend to be a key to the diagnosis. As detailed in Organophosphate Toxicity, one should keep in mind the mnemonics SLUDGE (salivation, lacrimation, urination, diarrhea, GI upset, emesis) and DUMBELS (diaphoresis and diarrhea; urination; miosis; bradycardia, bronchospasm, bronchorrhea; emesis; excess lacrimation; and salivation). [77]
As acetylcholine levels accumulate in the peripheral, autonomic, and central nervous systems, the effects can be variable and fluctuating:
-
Bradycardia, hypotension, and miosis from muscarinic effects and tachycardia, hypertension, and mydriasis from nicotinic effect can occur.
-
Excess acetylcholine at the nerve muscle junction can cause fasciculations and cramps, but then when the depolarization block becomes predominant, weakness and flaccidity are evident.
-
Any muscle or muscle group can be affected. Typically, neck muscles, cranial nerve muscles, respiratory muscles, and proximal limb muscles are the hardest hit. One of the significant points to remember is that, in the same patient, some muscles may be virtually unaffected, others mildly, and others severely affected. This is also true when myasthenic patients are treated with anti-acetylcholinesterases such as pyridostigmine. Sometimes, certain muscles in the same patient may still be weak from the myasthenia at the same time that other muscles are overdosed on the pyridostigmine.
-
Central effects may include mixtures of anxiety and emotional lability, ataxia, delirium, coma, and epileptic seizures.
It is often useful to divide organophosphate poisoning into stages in which initially, in addition to the muscle paralysis, the immediate cholinergic effects of salivation, lacrimation, urination, diarrhea, GI upset, emesis, diaphoresis, miosis (sometimes alternating with mydriasis), bradycardia (sometimes alternating with tachycardia), bronchospasm, and bronchorrhea predominate. A variable degree of mental effects from anxiety to severe delirium may be present. Then, an "intermediate syndrome" dominated by muscle paralysis alone, especially the proximal and respiratory muscles, occurs. [78, 79] However, the initial symptoms may still persist in various degrees. Finally weeks or months later, peripheral neuropathies, muscle necrosis (presumably from overstimulation), CNS damage (perhaps resulting from impaired respiration and circulation but perhaps direct effects of cholinergic overdose), and other long-term sequelae may occur. Some of the sequelae may be partially reversible overtime. [80]
As is the case for botulism, blood tests for anti-acetylcholinesterase poisoning present a problem, considering the need for rapid diagnosis. One approach is to do an enzyme activity assay for cholinesterases. Although this has been automated recently using robotic techniques, one still needs to send the blood to a high-level laboratory and wait for processing. [81] Another approach is to detect the organophosphate compound. This also presents difficulties including the question of which organophosphate compounds are detected.
Recently, various rapid methods have been developed including biosensors, which use genetically engineered bacteria whose membranes contain enzymes capable of breaking down certain organophosphates into more easily identified compounds. [82] In most hospital situations, the blood would have to be sent to a highly specialized reference laboratory. Thus, the neurophysiologic techniques, though imperfect, can be especially useful for rapidly deciding whether or not individual or multiple cases of paralysis might involve poisoning with an anti-acetylcholinesterase.
Far fewer cases of acetylcholinesterase toxicity have been studied neurophysiologically than is the case for myasthenia gravis. Many of the cases that were studied were examined under emergency conditions of mass intoxications and, thus, could not be studied as fully as is the situation for myasthenia gravis. Therefore, the significance of the findings are less certain. In this, as in most other situations, combining neurophysiologic findings with information from the history and physical examination is best.
The following is a summary of the main neurophysiologic findings that have been seen in cases of anti-acetylcholinesterase intoxication:
-
A single supramaximal electrical stimulation induces repetitive responses. This tends to occur at the earliest stage. It can also occur in slow channel syndrome.
-
A decremental response to repetitive stimulation at low frequency (2 or 3 Hz) with lack of posttetanic or postexercise facilitation. The decrement may be absent. In any case, this would not distinguish from MG, LEMS, or botulism. Lack of a period of posttetanic (or postexercise) facilitation (ie, temporary increase in strength after maximal or near-maximal muscle activation) is against myasthenia and LEMS.
-
A decrement-increment response to high-frequency stimulation (usually 30-50 Hz). One group has found that the decrement-increment response is more common either in the early, before the greatest severity is reached, or in the late stages after the worst has passed. [83, 84] Fasciculations are also commonly seen in these stages.
Neuromuscular Transmission and Terrorism
Blocking neuromuscular transmission is a potential mode of terrorism. Neuromuscularly oriented clinicians should think about how they might use the neuromuscular assessment techniques to help combat this threat.
Botulinum toxin and anti-acetylcholinesterases (organophosphates) are probably the two toxins that are the most likely to be used because they could be made available in sufficient amounts. In response to a possible terrorist attack with such agents, the principles and methods of neuromuscular transmission assessment described above could prove helpful in the initial as well as later stages of victim assessment.
As in medical cases, the initial history and clinical evaluation of the patients takes precedence.
For botulism, consider contaminated food, wounds, or marks indicating injections, and the possibility of toxin inhalation. On physical examination, the previously mentioned “dozen D’s” may be helpful: “dry mouth, diplopia, dilated pupils, droopy eyes, droopy face, diminished gag reflex, dysphagia, dysarthria, dysphonia, difficulty lifting head, descending paralysis, and diaphragmatic paralysis.” [70]
For organophosphate poisoning, some independent evidence that it might have been a gas attack would be suggestive. Organophosphates act in minutes, so the context would be people suddenly stricken in a relatively confined area, such as a subway platform. In contrast, botulism has an onset on the order of hours to days. One must also consider that the purified botulinum toxin could be dispersed as an aerosol. Still, some time lag would exist, depending on how much toxin was delivered. For example, in toxin injected for treatment of blepharospasm, the patient usually does not notice much of an effect until the next day. However, extremely high doses could work much more quickly.
The previously mentioned mnemonics SLUDGE (salivation, lacrimation, urination, diarrhea, GI upset, emesis) and DUMBELS (diaphoresis and diarrhea; urination; miosis; bradycardia, bronchospasm, bronchorrhea; emesis; excess lacrimation; and salivation) could be helpful. However, one must consider that many of the signs can fluctuate in organophosphate poisoning: bradycardia, hypotension, and miosis from muscarinic effects and tachycardia, hypertension, and mydriasis from nicotinic effects. Excess acetylcholine can cause fasciculations and cramps, but, later when depolarization block sets in, weakness and flaccidity will predominate. Neck muscles, cranial nerve muscles, respiratory muscles, and proximal limb muscles often are the most severely affected. Central effects can include anxiety and emotional lability, ataxia, delirium, coma, and epileptic seizures. In addition, recall the 3 stages of acute paralysis, intermediate syndrome, and later sequelae.
Prakash and Lo have discussed a simple framework for distinguishing between organophosphate poisoning (ie, anti-acetylcholinesterase, botulinum toxin) and a group consisting of either anthrax or cyanide. [83]
First, standard nerve conductions with supramaximal stimulation should be performed. The compound motor action potential (CMAP) amplitudes should be observed. If they are normal, then the poisoning is likely in the anthrax/cyanide group. If the CMAP amplitude is reduced, then the poisoning is either organophosphate or botulism.
For the reduced CMAP group, repetitive stimulation is now performed. (1) If no decrement is present at 3 Hz but either a decrement-increment pattern or a decrement pattern at 30-50 Hz, then one should suspect organophosphate poisoning. (2) If a decrement is present at 3 Hz and an increment at 30-50 Hz, then one should suspect botulinum toxin poisoning. [88]
Additional confirmation might be obtained by single-fiber EMG. Both organophosphate poisoning and botulism should show increased jitter and blocking. However, with increased firing rates, the jitter and blocking in botulism often (but not always) ameliorates, whereas it does not in organophosphate poisoning. Careful attention to divergent autonomic features is helpful. For example, patients with botulism have dilated pupils (which constrict to light poorly) and dry mouth. Organophosphate toxicity causes pupillary constriction (in some phases of the toxicity) and excess salivation. One must always remember that not all potential findings are present in every patient and findings can fluctuate over time. No absolutely foolproof schema that is guaranteed to work in every situation exists. A good knowledge and understanding of neuromuscular assessment techniques should be helpful if the need for rapid triage arises.
Tables
Both for the more normal clinical purposes and also to aid in differentiating the causes of weakness in large-scale poisonings and possible terrorist situations, the basic points made in this article are summarized in the tables below. Table 1 is a summary of the overall clinical assessment information, and Table 2 summarizes the neurophysiologic assessment methods.
Table 1. Clinical Assessment of Neuromuscular Transmission (Open Table in a new window)
Disease or Toxicity |
History |
Time of Onset |
Method of Transmission or Mechanism |
Clinical Findings |
Pharma-cologic Testing |
Immuno-logical and Other Blood Tests |
Neuro-physiologic Tests |
MG |
Fatigability, weakness toward the end of the day, diplopia, ptosis, difficulty swallowing or other bulbar signs |
Traditionally, young women and older men. More older than younger patients now. Can be gradual or rapid onset. |
Most appear to be sporadic due to antibodies to AChR or related muscle membrane proteins such as MUSK. |
Cranial nerve weakness, eg, ptosis, diplopia Difficulty with repetitive motions or holding fixed postures Clinical tests: Simpson test Cogan lid twitch sign Extended fatigue and recovery test Enhanced ptosis test Ice pack test |
Tensilon Neo-stigmine Pyridostig-mine |
Antibodies to AChR, MUSK, LRP4 |
EMG Nerve conduction studies Repetitive stimulation Single-fiber EMG |
Congenital myasthenia |
Generally similar to noncongenital myasthenia. See Congenital (Genetic) Myasthenic Syndromes for more details. |
Usually gradual onset from birth or early age; some later |
Genetic: Mutation often in AChR, AchE, or ChAT Possibly also mutations in synaptobrevin and other molecules |
Generally similar to non-congenital myasthenia |
Some forms improve with Tensilon (not the types with mutant AchE). |
Gene sequence available for some |
Mouse bioassay is definitive test. ELISA is available but not as definitive. Endopep-MS was recently developed. |
LEMS |
Insidiously developing weakness |
Usually in middle to later adult life Frequently gradual onset |
Patients develop antibodies to presynaptic VGCCs, often as a side effect of cancer (paraneoplastic). |
Weakness is greater in legs and proximal muscles. Usually no bulbar or ocular weakness present. Weakness may ameliorate with repetition. |
Tensilon test result is usually negative, but negative response not evidence for LEMS. |
Antibodies to VGCCs, P/Q type |
Mouse bioassay is definitive test. ELISA is available but not as definitive. Endopep-MS was recently developed. |
Botulism |
Rapidly progressive weakness and autonomic dysfunction |
Hours to days from ingestion |
Infection may come from food or wounds. Purified toxins may be deliberately injected into victims or sprayed into the air for inhalational poisoning |
"Dozen D's" [70] : "dry mouth, diplopia, dilated pupils, droopy eyes, droopy face, diminished gag reflex, dysphagia, dysarthria, dysphonia, difficulty lifting head, descending paralysis, and diaphra-gmatic paralysis" |
Tensilon test result may be positive. |
Mouse bioassay is definitive test. ELISA available but not as definitive. Endopep-MS was recently developed. |
Mouse bioassay is definitive test. ELISA is available but not as definitive. Endopep-MS was recently developed. |
Anti-acetylchol-inesterase toxicity |
Rapidly develop weakness, autonomic dysfunction, and CNS dysfunction |
Minutes from exposure |
Accidental ingestion of pesticides Deliberate poisoning for military or terrorist purposes (saran or soman) Overtreatment with pyridostigmine |
Muscarinic - Bradycardia, hypotension, and miosis Nicotinic - Tachycardia, hypertension, and mydriasis Muscles - Fascicu-lations and cramps progressing to weakness and flaccidity Central effects - Anxiety, emotional lability, ataxia, delirium, coma, and epileptic seizures Mnemonics - SLUDGE and DUMBELS |
Tensilon test should do nothing or make patient worse. |
Chemical and biosensor techniques to detect acetylcholinest-erase activity and organo-phosphates exist but are not readily available. |
EMG Nerve conduction studies Repetitive stimulation Single-fiber EMG |
AChR: Acetylcholine receptor
AchE: Acetylcholinesterase
ChAT: Cholineacetyltransferase
CNS: Central nervous system
MUSK: Muscle-specific tyrosine kinase
LEMS: Lambert-Eaton myasthenic syndrome
VGCCs: Voltage-gated calcium channels
Endopep-MS: Endopeptidase cleavage mass spectroscopy
ELISA: Enzyme-linked immunosorbent assay
SLUDGE: Salivation, lacrimation, urination, diarrhea, GI upset, emesis
DUMBELS: Diaphoresis and diarrhea; urination; miosis; bradycardia, bronchospasm, bronchorrhea; emesis; excess lacrimation; and salivation.
Table 2. Neurophysiologic Assessment of Neuromuscular Transmission (Open Table in a new window)
Disease or Toxicity |
CMAP Amplitude |
Repetitive Stimulation (Slow) |
Repetitive Stimulation (Fast) |
Effect of Exercise or Tetanic Stimulation on Subsequent Repetitive Stimulation |
Repetitive CMAP With Single Stimulation |
EMG Fibs/PSWs MUAP |
SFEMG |
MG |
Normal |
Decrement common (2-10Hz) |
At >10Hz, can increase (not reliable for diagnosis of MG) |
Initially, 10-30 sec contraction or high-frequency (tetanic) stimulation repairs decrement for up to 2 min. Then, postexercise exhaustion occurs for 2-5 min. Then, return to original response. |
Not seen |
No Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude |
Increased jitter or blocking |
Congenital myasthenia |
Normal |
Decrement common |
Variable, not useful for diagnosis |
Generally same as for MG above |
Occurs in some types, eg, endplate AChE deficiency and slow channel syndrome |
No Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking |
LEMS |
Greatly reduced |
Decrement common |
Significant increment, useful for diagnosis |
Initially, 10 sec contraction or high-frequency (tetanic) stim repairs decrement for 20-30 sec and also greatly increases CMAP size (50-100%). Then, postexercise exhaustion occurs for 2-5 min. Then, return to original response. |
Not seen |
No Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking |
Botulism |
Reduced but usually less than LEMS |
Decrement common |
Increment, but usually less than LEMS |
Often similar to LEMS but much less robust response (smaller increase). The small increase may last longer (a few min) before return to prior size. Also, no postexercise exhaustion occurs. |
Not seen |
Sometimes Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking, which may decrease as the firing rate increases. |
Anti-acetylcho-linesterase toxicity |
May be reduced but usually not severely |
Decrement may occur. |
Decrement-increment pattern in relatively mild weakness. Decrement in severe weakness. |
No postexercise facilitation |
Often |
Often has Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking |
CMAP: Compound motor action potential
LEMS: Lambert-Eaton myasthenic syndrome
Fibs: Fibrillations
PSWs: Positive sharp waves
MUAP: Motor unit action potential
Stim: Stimulation
MG: Myasthenia gravis
-
A typical decrementing response to repetitive nerve stimulation in myasthenia gravis. The amplitude of the initial response is normal, and the decrement is maximal in the fourth response. Thereafter, the responses increase somewhat, giving a U-shaped envelope to the train of responses.
-
A diagram of pseudofacilitation in the muscle responses seen during high-frequency repetitive nerve stimulation. Amplitude of the responses increases due to shortening of the waveforms, but the area under the waveforms remains relatively constant.
-
Typical activation cycles seen during repetitive nerve stimulation. The vertical lines represent the amplitude of muscle responses during 3-Hz repetitive nerve stimulation before and at indicated intervals after activation of the muscle by brief maximum voluntary contraction (arrow). In myasthenia gravis (MG), a decrementing response is observed initially, which partially repairs after activation and is later exaggerated. In Lambert-Eaton myasthenic syndrome (LEMS), the responses are small. A decrementing response occurs, and activation is followed by a marked and brief increase in response amplitude.
-
Electrode positions for performing repetitive nerve stimulation of the trapezius muscle. The active recording electrode (black) is placed over the belly of the muscle and the reference-recording electrode (red) distally over the shoulder. The spinal accessory nerve is stimulated as it crosses the sternocleidomastoid muscle. The green wire is the ground.
-
Action potentials from a single motor unit in a patient with Lambert-Eaton myasthenic syndrome. The waveforms vary from discharge to discharge, indicating abnormal neuromuscular transmission.
-
Repetitive nerve stimulation studies in Lambert-Eaton myasthenic syndrome. The initial response amplitude is low, and a decrementing response to low-frequency stimulation is noted (A). Immediately after activation, the amplitude is increased (B). During high-frequency stimulation (C), the initial decrement is followed by an increase in amplitude to more than twice the initial value.
-
Muscle responses to nerve stimulation in a patient with Lambert-Eaton myasthenic syndrome. The initial response (left) is very small. After brief maximum voluntary contraction, the amplitude (right) increases markedly.
-
Muscle responses to 3-Hz nerve stimulation in a hand muscle of a patient with familial infantile myasthenia. The initial response (A) is normal, but a decrementing response occurs after prolonged 3-Hz stimulation (B).
-
Responses to 3-Hz nerve stimulation in a hand muscle in a patient with slow channel syndrome. A decrementing pattern is noted, and a repetitive discharge (black) is seen following the response to the initial stimulus.
-
Repetitive nerve stimulation study. The baseline is unstable, causing the compound muscle action potential (CMAP) waveforms to move up and down, giving an irregular envelope to the train. Muscle movement is the most likely cause.
-
This figure shows a decremental response to repetitive nerve stimulation in the deltoid muscle of a patient with a severe cervical radiculopathy.
-
Repetitive nerve stimulation test in a hand muscle.
Tables
Disease or Toxicity |
History |
Time of Onset |
Method of Transmission or Mechanism |
Clinical Findings |
Pharma-cologic Testing |
Immuno-logical and Other Blood Tests |
Neuro-physiologic Tests |
MG |
Fatigability, weakness toward the end of the day, diplopia, ptosis, difficulty swallowing or other bulbar signs |
Traditionally, young women and older men. More older than younger patients now. Can be gradual or rapid onset. |
Most appear to be sporadic due to antibodies to AChR or related muscle membrane proteins such as MUSK. |
Cranial nerve weakness, eg, ptosis, diplopia Difficulty with repetitive motions or holding fixed postures Clinical tests: Simpson test Cogan lid twitch sign Extended fatigue and recovery test Enhanced ptosis test Ice pack test |
Tensilon Neo-stigmine Pyridostig-mine |
Antibodies to AChR, MUSK, LRP4 |
EMG Nerve conduction studies Repetitive stimulation Single-fiber EMG |
Congenital myasthenia |
Generally similar to noncongenital myasthenia. See Congenital (Genetic) Myasthenic Syndromes for more details. |
Usually gradual onset from birth or early age; some later |
Genetic: Mutation often in AChR, AchE, or ChAT Possibly also mutations in synaptobrevin and other molecules |
Generally similar to non-congenital myasthenia |
Some forms improve with Tensilon (not the types with mutant AchE). |
Gene sequence available for some |
Mouse bioassay is definitive test. ELISA is available but not as definitive. Endopep-MS was recently developed. |
LEMS |
Insidiously developing weakness |
Usually in middle to later adult life Frequently gradual onset |
Patients develop antibodies to presynaptic VGCCs, often as a side effect of cancer (paraneoplastic). |
Weakness is greater in legs and proximal muscles. Usually no bulbar or ocular weakness present. Weakness may ameliorate with repetition. |
Tensilon test result is usually negative, but negative response not evidence for LEMS. |
Antibodies to VGCCs, P/Q type |
Mouse bioassay is definitive test. ELISA is available but not as definitive. Endopep-MS was recently developed. |
Botulism |
Rapidly progressive weakness and autonomic dysfunction |
Hours to days from ingestion |
Infection may come from food or wounds. Purified toxins may be deliberately injected into victims or sprayed into the air for inhalational poisoning |
"Dozen D's" [70] : "dry mouth, diplopia, dilated pupils, droopy eyes, droopy face, diminished gag reflex, dysphagia, dysarthria, dysphonia, difficulty lifting head, descending paralysis, and diaphra-gmatic paralysis" |
Tensilon test result may be positive. |
Mouse bioassay is definitive test. ELISA available but not as definitive. Endopep-MS was recently developed. |
Mouse bioassay is definitive test. ELISA is available but not as definitive. Endopep-MS was recently developed. |
Anti-acetylchol-inesterase toxicity |
Rapidly develop weakness, autonomic dysfunction, and CNS dysfunction |
Minutes from exposure |
Accidental ingestion of pesticides Deliberate poisoning for military or terrorist purposes (saran or soman) Overtreatment with pyridostigmine |
Muscarinic - Bradycardia, hypotension, and miosis Nicotinic - Tachycardia, hypertension, and mydriasis Muscles - Fascicu-lations and cramps progressing to weakness and flaccidity Central effects - Anxiety, emotional lability, ataxia, delirium, coma, and epileptic seizures Mnemonics - SLUDGE and DUMBELS |
Tensilon test should do nothing or make patient worse. |
Chemical and biosensor techniques to detect acetylcholinest-erase activity and organo-phosphates exist but are not readily available. |
EMG Nerve conduction studies Repetitive stimulation Single-fiber EMG |
Disease or Toxicity |
CMAP Amplitude |
Repetitive Stimulation (Slow) |
Repetitive Stimulation (Fast) |
Effect of Exercise or Tetanic Stimulation on Subsequent Repetitive Stimulation |
Repetitive CMAP With Single Stimulation |
EMG Fibs/PSWs MUAP |
SFEMG |
MG |
Normal |
Decrement common (2-10Hz) |
At >10Hz, can increase (not reliable for diagnosis of MG) |
Initially, 10-30 sec contraction or high-frequency (tetanic) stimulation repairs decrement for up to 2 min. Then, postexercise exhaustion occurs for 2-5 min. Then, return to original response. |
Not seen |
No Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude |
Increased jitter or blocking |
Congenital myasthenia |
Normal |
Decrement common |
Variable, not useful for diagnosis |
Generally same as for MG above |
Occurs in some types, eg, endplate AChE deficiency and slow channel syndrome |
No Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking |
LEMS |
Greatly reduced |
Decrement common |
Significant increment, useful for diagnosis |
Initially, 10 sec contraction or high-frequency (tetanic) stim repairs decrement for 20-30 sec and also greatly increases CMAP size (50-100%). Then, postexercise exhaustion occurs for 2-5 min. Then, return to original response. |
Not seen |
No Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking |
Botulism |
Reduced but usually less than LEMS |
Decrement common |
Increment, but usually less than LEMS |
Often similar to LEMS but much less robust response (smaller increase). The small increase may last longer (a few min) before return to prior size. Also, no postexercise exhaustion occurs. |
Not seen |
Sometimes Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking, which may decrease as the firing rate increases. |
Anti-acetylcho-linesterase toxicity |
May be reduced but usually not severely |
Decrement may occur. |
Decrement-increment pattern in relatively mild weakness. Decrement in severe weakness. |
No postexercise facilitation |
Often |
Often has Fibs/PSWs MUAPs may be unstable, polyphasic, brief, or low amplitude. |
Increased jitter or blocking |

What would you like to print?
- Overview
- Myasthenia Gravis: Clinical Evaluation
- Repetitive Nerve Stimulation
- Electromyography
- RNS Versus Single-Fiber EMG
- Lambert-Eaton Myasthenic Syndrome
- Congenital (Genetic) Myasthenic Syndromes
- Botulism
- Anti-Acetylcholinesterase Toxicity
- Neuromuscular Transmission and Terrorism
- Tables
- Show All
- Media Gallery
- Tables
- References





