Overview
The presentation of degenerative disease in focal areas of the cerebral cortex is the hallmark of the family of diseases referred to as frontotemporal dementia (also termed frontotemporal lobar degeneration). [1, 2] Cases of elderly patients with progressive language deterioration have been described since Arnold Pick's landmark case report of 1892. His case study "On the relationship between aphasia and senile atrophy of the brain" still serves as a frame of reference for apparently focal brain syndromes in diffuse or generalized degenerative diseases of the brain. [3] As Pick stated, “simple progressive brain atrophy can lead to symptoms of local disturbance through local accentuation of the diffuse process.”
In the 1980s and 1990s, two parallel streams of information accumulated related to focal brain degenerations. In 1982, Mesulam reported 6 patients with progressive aphasia, gradually worsening over a number of years, who did not develop a more generalized dementia. [4] Since Mesulam's publication, numerous other cases have been reported, and Mesulam’s group has contributed additional reviews. [5, 6] This disorder, which is currently termed primary progressive aphasia (PPA), has gained acceptance as a syndrome.
Subsequently, the PPA syndrome was defined as a disorder limited to progressive aphasia, without general cognitive impairment or dementia, over a 2-year period. [5] Many patients develop more generalized dementia later in the course of the illness, as reported by Kirshner et al. [7] Less commonly, cases of isolated right frontal or temporal degeneration have been reported. [8, 9] These patients experience failure to recognize family members (prosopagnosia), failure to remember topographic relationships, and similar disorders. (See the image below.)
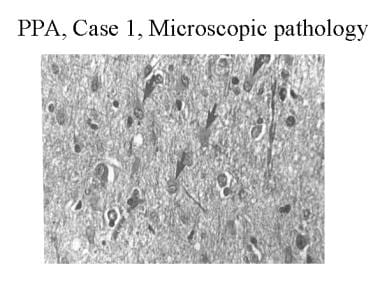 Hematoxylin and eosin stain of the left frontal cortex from a patient with primary progressive aphasia. This shows loss of neurons, plump astrocytes (arrow), and microvacuolation of the superficial cortical layers. Reproduced with permission of John Wiley & Sons, Inc.
Hematoxylin and eosin stain of the left frontal cortex from a patient with primary progressive aphasia. This shows loss of neurons, plump astrocytes (arrow), and microvacuolation of the superficial cortical layers. Reproduced with permission of John Wiley & Sons, Inc.
In England and Europe, cases of frontal lobe dementia were described with progressive dysfunction of the frontal lobes. In a series of case reports, Neary and Snowden outlined a syndrome with initial symptoms that were suggestive of psychiatric illness. However, the following frontal lobe behavioral abnormalities appeared over time:
-
Disinhibition
-
Impulsivity
-
Impersistence
-
Inertia
-
Loss of social awareness
-
Neglect of personal hygiene
-
Mental rigidity, stereotyped behavior
-
Utilization behavior - Ie, a tendency to pick up and manipulate any object in the environment
These descriptions included language abnormalities such as reduced speech output, mutism, echolalia, and perseveration.
The condition described in the North American literature as primary progressive aphasia and that described in the European literature as frontal dementia have been combined under the term frontotemporal lobe dementia (FTD) or frontotemporal lobar degeneration (FTLD). Within this grouping, the frontal lobe syndrome described by Neary and Snowdon, [10, 11] is referred to as, interchangeably, behavioral variant frontotemporal lobe dementia (bvFTD) or frontal variant frontotemporal lobe dementia (fvFTD). The progressive aphasias have been divided into 3 groups: progressive nonfluent aphasia, semantic dementia, and logopenic progressive aphasia. [12, 13, 14]
In recent years, the term frontotemporal dementia has become an umbrella term referring to clinical syndromes of frontal dementia or progressive aphasia. An alternate term, frontotemporal lobar degeneration, relates to pathologies associated with the frontotemporal lobe dementia syndromes. In this review, the 2 terms are used more or less synonymously.
Treatment
To date, most efforts have concentrated on diagnosing FTD and understanding its pathogenesis. Once the abnormal gene products are better understood, specific medical treatments may emerge. At present, however, medical treatment is extremely limited.
Social interventions, counseling, and speech/language/cognitive therapy to facilitate the use of spared functions may make the condition easier to bear for the patient, caregivers, and family members. Whether behavioral interventions slow the progression of the disease remains to be proved.
All current pharmacologic treatments are unproved, but selective serotonin reuptake inhibitor (SSRI) antidepressants and trazodone are widely recommended. [14, 15, 16] Cholinesterase inhibitors, approved for Alzheimer disease, are sometimes used in this condition, but there is no evidence that FTD involves a cholinergic deficit, and there is no clinical evidence of benefit. [17] Anecdotally, some patients may improve in terms of memory, but others seem to worsen in behavior. Likewise, the drug memantine (Namenda) has been used in FTD, but 2 recent small clinical trials failed to confirm any benefit. [18, 19] It is hoped that future breakthroughs in the molecular biology and genetics of these disorders may lead to disease-modifying treatments.
Etiology
Considerable progress has been made with regard to understanding the genetics and molecular pathology of frontotemporal lobe dementia (FTD). Prior to the late 1990s, the FTD syndromes were defined by clinical syndromes and by light microscopic histopathology only. FTD cases included Pick disease and non-Pick lobar atrophy cases lacking the silver staining Pick bodies in neurons. This pathology was referred to as "dementia lacking specific histologic features. [20] Kirshner and colleagues also described 2 cases of primary progressive aphasia (PPA) in which autopsy documented only focal neuronal loss, gliosis, and microvacuolation of the cortex, changes resembling a nonspecific dementia or FTD. [21]
Tau protein
In 1998, Hutton and colleagues [22] and Poorkaj and colleagues [23] described mutations in the microtubule-associated protein tau (MAPT) gene, located on chromosome 17, associated with FTD syndromes and insoluble tau deposits. FTD consequently changed from a lobar atrophy of syndromic definition to a group of molecular genetic disorders. Many MAPT mutations have subsequently been described, including the 2 large families reported by Morris and colleagues, under the term “hereditary dysphasic dementia.” [24]
Tau is the major protein component of Pick bodies and is seen in a number of other neurodegenerative diseases, including progressive supranuclear palsy (PSP), [25] corticobasal degeneration, and the amyotrophic lateral sclerosis (ALS) ̶ Parkinson dementia disease complex of Guam. These other diseases can produce syndromes of primary language degeneration resembling PPA and FTD. Interestingly, no MAPT mutations have been found in Alzheimer disease.
FTLD-U, Progranulin mutations, TDP-43, and FUS accumulation
Until the past few years, more than 50% of cases of FTD, even the familial ones, were not associated with tau pathology, although many of these cases were also linked to chromosome 17. Most such cases have been noted to have ubiquitin immunoreactive inclusions in the cytoplasm or nucleus or ubiquitin immunoreactive neurites. This group has been designated frontotemporal lobar degeneration-ubiquitin (FTLD-U), [26] though as we shall see, this term is being replaced by reference to specific mutations.
In 2006, 2 teams of investigators reported mutations in the progranulin gene on chromosome 17 as the cause of this syndrome. [27, 28] Many such cases have now been reported, with mutations resulting in a premature termination codon, causing haploinsufficiency. The progranulin mutations result in a loss of protein, whereas the tau mutations result in a toxic gain of function. The TAR ̶ deoxyribonucleic acid (DNA) binding protein (TDP-43) is a major component of the ubiquitinated inclusions in most of these cases. Progranulin mutations and the TDP-43 positive inclusions have been found in cases of FTD, PPA, and corticobasal degeneration. [29, 30]
Other mutations
Tau and progranulin mutations appear to account for the most cases of FTD, and at least in familial cases, true examples of dementia lacking distinctive histologic features have become much less common. Moreover, other gene loci have been implicated in this family of disorders. Cases of inclusion body myopathy with Paget disease of the bone associated with FTD have been reported and have been associated with mutations involving chromosome 9, the valosin-containing protein (VCP) gene. [31, 32] In some of these cases, TDP-43 protein accumulation has been reported.
Other mutations on chromosome 9 have been associated with FTD-ALS. [33] The most common of these is the C9ORF72 gene, in which hexanucleotide repeat expansions have been reported. This mutation can be seen in patients with FTD, usually the behavioral variant, or familial ALS, or both. [34, 35, 36, 37] Finally, mutations of the chromatin-modifying protein 2B (CHMP2B) gene on chromosome 3 have also been reported in a Danish family. These mutations have generally not been associated with TDP-43 protein accumulation. [38]
Finally, there are a few FTD cases with ubiquitin deposition, but no TDP-43 accumulation, in whom both tau and progranulin mutations are absent. Rather, an accumulation of “fused in sarcoma” (FUS) proteins have been found. Reported cases have included both FTD and ALS. [39, 40]
Alzheimer disease and FTD
Alzheimer disease, by far the most common dementing pathology, has also been associated with syndromes of PPA and FTD in some cases. [7, 41, 42, 43] Alzheimer disease is uncommonly found at autopsy in patients with primary progressive, nonfluent aphasia, but the association is more common in semantic dementia and is especially frequent in patients with logopenic progressive aphasia. The relationship of Alzheimer disease to the primary progressive aphasia syndromes is covered in more detail in the section on Primary Progressive Aphasia. In addition, there is now a recognized syndrome of “frontal variant Alzheimer disease."
ALS and FTD
As mentioned above, patients with amyotrophic lateral sclerosis (ALS) develop behavioral and cognitive changes, often consistent with behavioral-variant FTD. Most such cases have ubiquitin-immunoreactive inclusions found in the frontal cortex, and these have been associated with the C9ORF72 gene mutation. About 10% of patients with ALS have significant dysfunction in behavior and executive function, and some patients with FTD develop motor neuron disease as their illness progresses. [34, 37]
Genetic Distribution and Variation
The cause of frontotemporal lobe dementia (FTD) is unknown, but, as discussed earlier, significant evidence supports a genetic component to these syndromes. [44]
As many as 40–50% of patients with FTD have an affected family member. In a Dutch study, 38% of the index cases of FTD had a first-degree relative with similar symptoms at an early age of onset. A review by Seelaar and colleagues found that 27% of cases had autosomal-dominant inheritance. [45]
The molecular genetics of FTD has become much more complex in recent years. As mentioned above, the first genetic link in FTD was to markers on band 17q21-22, the gene locus for the tau protein.
The tau gene marker has linked cases of FTD in several Dutch families, cases of hereditary dysphasic dementia reported in the United States, and a variety of other clinical syndromes called tauopathies, including familial parkinsonism with dementia, corticobasal degeneration, Pick disease without Pick bodies, and progressive supranuclear palsy (PSP). Overlap cases with these other tauopathies have been reported. The pathophysiology involves abnormal tau proteins, leading to the designation of FTD as one of a series of tauopathies.
In the words of an editorial by Wilhelmsen, the linkage of FTD with this gene site has put frontotemporal lobe dementia "on the map" (ie, gene map). [46] Many cases of familial FTD with specific tau mutations have now been reported. On the other hand, apolipoprotein E4 (APOE-4), which increases the risk for late-onset, sporadic Alzheimer disease, does not appear to have increased frequency in patients with FTD or primary progressive aphasia (PPA). [47, 48]
In the published series by Seelaar et al of 364 patients with FTD, 27% had positive family histories, which suggests autosomal dominant inheritance. Of these, 11% had tauopathies secondary to mutations of the MAPT gene on chromosome 17, whereas 6% had progranulin mutations. [49] These authors also reported that 10% had autosomal dominant inheritance patterns without a specific, demonstrated molecular genetic abnormality, adding up to 27% with autosomal dominantly inherited disorders. The other 73% of cases in their series were not clearly hereditary, and the molecular genetic basis of these disorders is not yet understood. [49]
In a National Institutes of Health study of 1425 FTD patients, the C9ORF72 mutation, with a pathogenic hexanucleotide repeat expansion, was associated with a large proportion of both sporadic and familial FTD cases. Specifically, the mutation was carried by 6% of white Europeans with sporadic FTD and 24.8% of white Europeans with familial FTD. The mutation was not found in Asian patients with sporadic FTD. [50]
Formerly, patients with all of these pathologies were lumped together under the term dementia lacking specific histological features, or nonspecific dementia. [20] Currently, most cases with autopsy study can be placed either in the tauopathy or the ubiquitin categories, so only rare cases are truly nonspecific. [51]
Epidemiology
Occurrence in the United States
The exact prevalence of frontotemporal lobe dementia (FTD) is unknown. Among patients presenting with dementia who are younger than 65 years, the prevalence may be similar to or greater than that of Alzheimer disease. [52, 53] Some series based on brain pathology have estimated that FTD is responsible for as many as 10%–20% of cases of dementia. [54] In the United States, estimates are generally lower; FTD ranks after Alzheimer disease, vascular dementia, and Lewy body dementia in frequency of dementing illnesses.
International occurrence
Studies from Lund, Sweden and Manchester, England estimated that FTD accounts for approximately 8% of patients with dementia. Probably the most accurate information comes from the Dutch study by Stevens et al, cited earlier, who reported 74 cases in a population of 15 million (ie, 5 cases per million persons). Among individuals aged 60-70 years, the prevalence was 28 cases per 100,000. [45]
Sex- and age-related demographics
FTD can develop at almost any age in either gender. A large case series compiled by Westbury and Bub investigated 112 published cases prior to 1997; males and females accounted for 66% and 34% of the cases, respectively. [52]
Most patients with FTD present in their 50s or 60s. In the review by Westbury and Bub, the mean age of onset was 59 years; the median age was 64 years. [52]
Prognosis
Frontotemporal lobe dementia (FTD), like all dementing illnesses, shortens life expectancy. The exact influence on mortality is unknown, and the rate of disease progression is variable.
Among patients younger than 65 years, FTD has a similar or greater prevalence, as compared with Alzheimer disease. Among patients in their 70s and older, the prevalence of Alzheimer disease far exceeds that of FTD. The average age of onset for FTD, as reported by Westbury and Bub, [52] is younger than that of Alzheimer disease.
The rate of progression from focal presentation to a more generalized dementia varies. The literature contains many cases of slowly progressive language dysfunction developing over a period of as long as 10-12 years, without obvious deterioration of other cognitive functions that would justify a diagnosis of dementia. Other patients progress to dementia within a few years.
One area of controversy in PPA concerns whether a generalized dementia eventually develops in all patients with PPA. The incidence of dementia in patients with PPA is unknown, but it likely approaches 50% over several years.
In the subset of cases of patients with FTD who develop motor neuron disease, the mortality rate is higher than for other FTD patients. Swallowing difficulty and aspiration pneumonia are especially common in this subgroup, but even patients with primary progressive aphasia (PPA) can develop dysphagia late in the course of the illness.
Primary Progressive Aphasia
For the subgroup of patients with frontotemporal lobe dementia (FTD) who have primary progressive aphasia (PPA), the presenting symptoms involve a deterioration of language function. [5, 6, 55] At first, other aspects of cognitive function and behavior may seem entirely normal. Pick first described the presentation of focal language deterioration as a sign of a dementing illness. [3] Sporadic cases were presented into the 20th century, until Mesulam named the syndrome of PPA. [4] Other authors, such as Kirshner et al [7] and Green et al, [41] described focal, aphasic presentations in patients who later showed signs of more general dementia.
Patients with PPA who do not depend on their verbal skills for their livelihood may continue to function at work. They do not act forgetful, they remember their way to familiar destinations, and they generally comport themselves normally, although some show the behavioral changes described under FTD. Artistic expressions may even increase or be taken on as new hobbies in these patients, [56, 57] although, according to Miller and Hou, [57] their productions are often compulsive in style.
The most common presenting symptom is word-finding difficulty. However, decreased fluency or hesitancy in producing speech, difficulty with language comprehension, and motor speech difficulties (eg, dysarthria) are also common. The descriptions of language syndromes in PPA have become more complex.
The mode of presentation in PPA suggests a focal lesion of the left hemisphere language cortex, but a focal lesion, other than evidence of focal atrophy, is usually not found. Magnetic resonance imaging (MRI), especially when combined with voxel-based morphometry, has become much more accurate in mapping localized areas of cortical atrophy. Rohrer and colleagues suggested that MAPT mutations are more often associated with symmetrical atrophy, whereas the progranulin mutations show more asymmetric atrophy. [58] There are also patterns of regional atrophy in the different FTD variants, which we will return to later. The course is progressive, with slowly worsening language function.
Initially, PPA was divided into 2 subtypes: (1) a progressive nonfluent aphasia, and (2) fluent aphasia with anomia. More recently, 3 subtypes have been described: (1) progressive nonfluent aphasia; (2) semantic dementia, and (3) logopenic progressive aphasia, also called the logopenic/phonologic variant. [12, 13]
Progressive nonfluent aphasia
In progressive nonfluent aphasia, speech is effortful and halting, with phoneme or speech sound errors; language production is simplified and agrammatic; and there is usually sparing of word comprehension and object knowledge, but often with impaired comprehension of syntax. This is, in other words, a Broca-like aphasia, often associated with dysarthria and with hesitant, groping speech and difficulty producing phonemes. These cases almost always have a non-Alzheimer pathology and most commonly a MAPT mutation or tau-based disorder. Rohrer and colleagues [59] reported that agrammatism and apraxia of speech were specifically useful in predicting the presence of a tau mutation and progressive nonfluent aphasia.
Semantic dementia
Semantic dementia is a fluent aphasia with impaired naming and impaired knowledge of word meanings, such that even single word comprehension becomes affected. Patients often have surface dyslexia in reading, spared repetition and motor speech, and, sometimes, poor object and person knowledge. Semantic dementia was first named by Snowden and colleagues [60] and was later defined by Hodges and his group in the United Kingdom. [61, 62] The pathology can be variable, including mostly FTLD-U cases, many with progranulin mutations, but a few have been associated with Alzheimer disease.
Logopenic progressive aphasia
The logopenic, or logopenic/phonologic, variant of PPA involves impaired naming and single word retrieval, impaired repetition of phrases and sentences, and, often, speech sound errors, with spared motor speech, spared single word comprehension and object knowledge, and absence of agrammatism. [63] This type of PPA is usually associated with a focal presentation of Alzheimer disease.
Behavioral Changes
In frontal lobe dementia or frontotemporal lobe dementia (FTD), as opposed to primary progressive aphasia (PPA), presenting symptoms often involve alterations in personality and social conduct. [10, 11, 64]
Patients with behavioral variant frontotemporal lobe dementia (bvFTD) may become disinhibited, developing a "witzelsucht," or fatuous sense of humor. Conversely, they may become apathetic, with little spontaneous speech or activity. They tend to neglect personal hygiene and to lose sensitivity to the effects of their behaviors on others.
Some develop frank frontal lobe behavioral abnormalities, such as hyperorality, utilization behavior (ie, picking up and manipulating any object in the environment, appropriate or not), and inappropriate sexuality.
Suggested criteria for the diagnosis of behavioral variant FTD have recently been published. [64] These are discussed below.
In the original descriptions of FTD, language function either is described as reduced in output (leading to muteness) or is characterized by perseveration, stereotyped responses, or even echolalia.
A relatively underappreciated symptom may be a sociopathic one and may first come to light in the context of breaking the law. [65]
Additional Presentations
A subgroup of patients with frontotemporal lobe dementia (FTD) develops signs and symptoms of motor neuron disease, such as fasciculations, muscle wasting and weakness, and bulbar symptoms. See above for the genetic basis of this syndrome.
Another subgroup of patients with FTD experiences progressive right hemisphere dysfunction. Why reports of progressive right hemisphere degeneration have been so much less common than those of progressive aphasia is unclear. [9]
Finally, a genetic disease with combined inclusion body myopathy and FTD has been described. [32]
Physical Examination
Physical and neurologic examinations reflect mainly the mental status abnormalities described under Behavioral Changes. Characteristics of frontotemporal lobe dementia (FTD), as found on physical examination, can be further described as follows:
-
Speech - Many patients have a nonfluent speech pattern, and virtually all have some degree of difficulty in naming or word finding
-
Behavior - Behavioral alterations and frontal lobe symptoms have been previously outlined
-
Ideation - Ideation tends to be concrete, with poor abstraction and organization of responses and delayed shifting of cognitive sets
-
Visual and spatial functions and constructional tasks – These are much less affected, except as influenced by behavioral and organizational difficulties; motor skills usually are spared, except for perseverative or inattentive responses and difficulty with temporal sequencing of tasks
-
Specific ideomotor apraxia – Rare, except in patients with language difficulty associated with corticobasal degeneration (see below).
-
Memory - Memory is usually preserved for orientation, although information retrieval may be difficult. Short-term memory deficits may be present in some patients but are less characteristic and early than those associated with Alzheimer disease.
-
Frontal release signs - Frontal release signs, such as a positive glabellar sign, snout, grasp, and palmomental responses, may develop
In a minority of patients, extrapyramidal signs, such as rigidity or even a full-blown parkinsonian syndrome of rigidity, akinesia, and tremor, may develop. These cases may overlap with Lewy body dementia, in which fluctuating mental status, early visual hallucinations, and REM sleep behavior disorder (acting out of dreams) are characteristic, along with mild parkinsonian motor symptoms.
An overlap also exists with the syndrome of corticobasal degeneration, in which rigidity and apraxia of the upper limbs, typically starting in one arm and remaining asymmetric, may coexist with neurobehavioral symptoms much like those associated with the syndrome of primary progressive aphasia (PPA). [66, 67, 68]
Widespread muscle atrophy, weakness, fasciculations, bulbar signs, and hyperreflexia may ensue in patients with motor neuron disease. Muscle weakness is also seen in the rare variant with inclusion body myopathy.
As mentioned earlier, some patients may show artistic or musical talents, sometimes with greater expression than before the onset of the illness.
Differential Diagnosis
Frontotemporal lobe dementia (FTD) is somewhat intermediate between focal disorders of the brain and more generalized neurodegenerative diseases.
The most important differential diagnoses for FTD involve focal pathologies such as brain tumors, abscesses, and strokes, as well as Alzheimer disease, which is a more common dementing illness than FTD.
In distinguishing between FTD and other focal lesions, gradually progressive onset, usually over years, is the key feature.
Brain imaging studies are helpful in ruling out focal, destructive, or neoplastic lesions. Whitwell and colleagues used voxel-based morphometry on MRI to distinguish differing patterns of lobar atrophy in variants of FTD with and without motor neuron disease. [69] See below under imaging for the contributions of MRI and positron emission tomography (PET) imaging to the diagnosis of FTD.
Alzheimer disease can mimic almost any of the FTD variants when it presents with focal symptoms. Only a few cases of pathologically confirmed Alzheimer disease have been reported that presented with isolated nonfluent aphasia, but more have been described with the syndromes of semantic dementia (although most cases of semantic dementia have the pathology of frontotemporal lobar degeneration-ubiquitin [FTLD-U]) and with the logopenic variant of primary progressive aphasia (PPA), which is most commonly associated with Alzheimer disease. Other “focal” presentations of Alzheimer disease include the “frontal variant” referred to earlier and the “posterior cortical atrophy” variant, in which visual symptoms predominate.
Kertesz et al suggested the term Pick complex to include the various non-Alzheimer pathologies, with or without Pick inclusion bodies and with or without motor neuron disease. [70] This terminology has not become widely adopted, so we will continue to use the clinical term frontotemporal lobe dementia and the pathologic term frontotemporal lobar degeneration.
Lab and EEG Studies
Routine testing (eg, blood, cerebrospinal fluid) in frontotemporal lobe dementia (FTD) is usually unrevealing.
The genetic test for APOE-4 is less useful in FTD than in Alzheimer disease. A study by Mesulam et al found no association between FTD and the APOE-4 genotype. [47] Other studies have had somewhat different results, but, in general, APOE-4 correlates much better with Alzheimer disease than with FTD. [48, 71]
The findings in electroencephalography (EEG) are commonly abnormal in FTD, often showing focal slowing of electrical activity over 1 or both frontal or temporal lobes. These findings are not sufficiently specific to be clinically useful, and, in general, EEG is less useful than functional brain imaging with PET scanning or even lobar atrophy on MRI.
Language and Neuropsychological Testing
Other than brain imaging studies, the most specific tests for evaluating frontotemporal lobe dementia (FTD) are evaluation with standardized language batteries and neuropsychological testing. [72, 73] Such studies assess the specific pattern of language abnormality and the presence of other cognitive and memory deficits. Preservation of many of these functions distinguishes FTD and primary progressive aphasia (PPA) syndromes from Alzheimer disease.
In distinguishing FTD from Alzheimer disease, the involvement of specific cognitive functions is the most important differentiating factor.
Grossman pointed to a double dissociation between immediate and short-term memory in a comparison study of 4 patients with PPA versus 25 patients with presumed Alzheimer disease. [73] Immediate memory was more impaired in PPA patients, whereas short-term memory deficits characterized the deficits of patients with Alzheimer disease. The frontal cortex, especially on the left side, is thought to be the site of working or immediate memory, whereas the hippocampus and other medial temporal structures, often affected early in Alzheimer disease, represent the site of short-term memory.
Other cognitive functions also showed differences between the 2 groups. Patients with PPA were more deficient than patients with Alzheimer disease on tasks of syntactic and speech fluency, correlating with the aphasia, and they had more severe impairment of attention (eg, digit span), another measure of immediate memory. Patients with PPA showed preserved memory and visuospatial functions, whereas those with Alzheimer disease were almost invariably impaired in these functions.
Atypical cases of Alzheimer disease can present with 3 “focal” syndromes: (1) aphasia, especially logopenic PPA, (2) a frontal syndrome resembling behavioral variant FTD, or (3)a visual-predominant form called posterior cortical atrophy. This issue of focal versus more generalized dementia, of course, harkens back to Pick’s original discussion of how neurodegenerative diseases progress.
Recent evidence supports neuron-to-neuron transmission of pathology in both Alzheimer disease and FTD, involving transmission of the abnormal misfolded proteins of tau or beta-amyloid from neuron to neuron in a way reminiscent of the prion diseases like Creutzfeldt-Jakob disease. [74] In other words, what eventually becomes a diffuse neurodegenerative disease has to start somewhere in the brain; it can start in one region of the brain and then spread, neuron to neuron, into unaffected areas.
PPA is a syndrome, not a pathological diagnosis. Although the term initially implied a pathology other than Alzheimer disease, we must now consider that some cases may have a syndrome of PPA but a pathological diagnosis of Alzheimer disease, or vice versa.
For example, in a series of 100 patients with focal presentations of dementing illness, 34 had autopsy-proved Alzheimer disease. [75] These cases comprised 12 (44.1%) of 26, patients with progressive nonfluent aphasia (PNFA). In comparison, only 2 (7.1%) of 28 of the behavioral-variant FTD patients and 2 (10%) of 20 with semantic dementia had postmortem evidence of Alzheimer disease.
Xiong and colleagues, [76] in a series of 33 autopsy studied cases of PNFA, 13 had Alzheimer disease pathology and 20 had FTD pathology. Four clinical features were useful in predicting FTD pathology: (1) age of onset before 60 years, (2) “sweet tooth”, or preference for sweet foods, (3) disinhibited behaviors, and (4) the presence of “knife-edge” atrophy in the frontal and/or temporal lobes. These features were only weakly predictive individually, but a majority of the FTD cases had at least one of the features, whereas most Alzheimer disease cases had none. Features that did not distinguish FTD and Alzheimer disease included progression to generalized dementia within 2 years (the defining characteristic in Mesulam’s original definition of PPA), presence of memory deficits (the usual presenting symptom in Alzheimer disease), or impairment of activities of daily living. In addition, onset before age 65 years did not distinguish cases of FTD and Alzheimer disease in thisseries, though the series had disproportionately young patients because of the authors’ interest in early dementia.
Hu and colleagues [77] reported 19 patients with PNFA and 19 with logopenic progressive aphasia. Twelve of the 19 logopenic progressive aphasia patients and 6 of 19 PNFA patients had either autopsy confirmation or CSF biomarkers suggestive of Alzheimer disease. Naming was more impaired in the Alzheimer disease patients, whereas letter-based fluency was more impaired in the non–Alzheimer disease cases.
According to revised guidelines from an international consortium, 3 of the following 6 clinically discriminating features indicate possible behavioral variant FTD (bvFTD):
Disinhibition
-
Apathy/inertia
-
Loss of sympathy/empathy
-
Preservative/compulsive behaviors
-
Hyperorality
-
Dysexecutive neuropsychological profile
Functional disability and characteristic neuroimaging should also be considered when diagnosing probable bvFTD. In addition, histopathologic evidence (brain biopsy or autopsy) or proof of a pathogenic mutation is needed for a diagnosis of bvFTD with definite frontotemporal lobar degeneration (FTLD). Commercial laboratories such as Athena now offer genetic testing for the tau, progranulin, and C9ORF72 mutations. The revised criteria improve diagnostic accuracy, particularly in the earliest stages. However, the authors cautioned that future studies are needed to establish reliability and specificity. [64]
Imaging Studies
Patterns of atrophy
Routine brain imaging with computed tomography (CT) scanning or MRI is usually remarkable only for cerebral atrophy. Some patients show impressive localized atrophy in the frontal or temporal lobe on 1 or both sides. (See the image below.). [78]
 Patient with progressive nonfluent aphasia. MRI showing focal, left temporal atrophy. Reprinted from Neurology in Clinical Practice, 4th ed. Kirshner H. Language and Speech Disorders. Copyright 2004, with permission from Elsevier.
Patient with progressive nonfluent aphasia. MRI showing focal, left temporal atrophy. Reprinted from Neurology in Clinical Practice, 4th ed. Kirshner H. Language and Speech Disorders. Copyright 2004, with permission from Elsevier.
On MRI, temporal lobe atrophy is especially easy to detect in the coronal projections. Cases differ as to the relative degree of atrophy in the frontal or temporal lobe and on the left versus right side. Research studies using voxel-based morphometry have provided more precise maps of the areas of focal atrophy.
Patients with frontal lobe neurobehavioral disorders (behavioral variant frontotemporal lobe dementia) often have bilateral frontal atrophy, especially involving the medial frontal cortex, sometimes with anterior temporal lobe atrophy as well.
Whitwell et al reported in 2006 that cases associated with motor neuron disease have more paracentral atrophy by voxel-based morphometry on MRI. [69]
Patients with PNFA tend to have perisylvian, left hemisphere atrophy, involving the frontal lobe and insula, sometimes extending into the temporal lobe.
Patients with semantic dementia typically have temporal lobe atrophy, often involving the anterior temporal lobes bilaterally. Patients with logopenic progressive aphasia often have bilateral superior temporal and inferior parietal atrophy. Patients with the related tauopathy progressive supranuclear palsy have midbrain tegmentum and superior cerebellar peduncle atrophy, and those with corticobasal degeneration have frontoparietal atrophy.
Hypometabolism
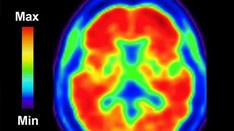
Functional imaging techniques, particularly single-photon emission computed tomography (SPECT) and positron emission tomography (PET) scanning, detect focal lobar hypometabolism or hypoperfusion with great sensitivity. (See the image below.). [79]
 Patient with progressive nonfluent aphasia. Positron emission tomography (PET) scan indicating hypometabolism of glucose in the left hemisphere. Reprinted from Neurology in Clinical Practice, 4th ed. Kirshner H. Language and Speech Disorders. Copyright 2004, with permission from Elsevier.
Patient with progressive nonfluent aphasia. Positron emission tomography (PET) scan indicating hypometabolism of glucose in the left hemisphere. Reprinted from Neurology in Clinical Practice, 4th ed. Kirshner H. Language and Speech Disorders. Copyright 2004, with permission from Elsevier.
The Hammersmith PET facility in London published early studies by Tyrell et al demonstrating that left temporal hypometabolism was observed in virtually all early cases of primary progressive aphasia (PPA). [80] More advanced cases also showed hypometabolism in the left frontal lobe and, occasionally, a lesser degree of hypometabolism in the right hemisphere.
These patterns of cortical involvement have been confirmed in many subsequent studies. The pattern of frontal and/or temporal involvement is distinct from that of Alzheimer disease, in which both parietal lobes tend to show the earliest hypometabolism.
New ligands used to bind to amyloid protein deposits (eg, Pittsburgh Compound B, or the recently approved florbetapir [Amyvid]) are helpful in the diagnosis of Alzheimer disease but not of FTD. These agents are approved by the FDA for safety but are not covered by Medicare or most private insurance policies.
Histologic Findings
Various pathologic findings have been reported in patients with primary progressive aphasia (PPA) and frontotemporal lobe dementia (FTD). The central theme of these reports is that these syndromes have a non-Alzheimer pathology. (See the image below.)
Hematoxylin and eosin stain of the left frontal cortex from a patient with primary progressive aphasia. This shows loss of neurons, plump astrocytes (arrow), and microvacuolation of the superficial cortical layers. Reproduced with permission of John Wiley & Sons, Inc.
Considering first the cases of PPA, Pick disease was the first pathologic disease associated with this syndrome. This was reported with a description of the language syndrome in 1892. The neuropathologic features of Pick disease are focal, lobar atrophy of the frontal and/or temporal lobes of 1 or both hemispheres, prominent gliosis associated with swollen neurons, and/or argentophilic inclusions (Pick bodies).
In the current era, several groups have reported cases of pathologically proven Pick disease. Holland et al, [81] Wechsler et al, [82] and Graff-Radford et al [83] have reported patients with pathologically proven Pick disease and progressive language deterioration. All patients described in these reports had slowly progressive language symptoms, with naming involved early. In all cases, enough cognitive functions were spared initially to make the disorder easily distinguishable from typical Alzheimer disease.
Many reported cases do not have Pick bodies but have the less specific findings of lobar atrophy, neuronal loss, gliosis, and microvacuolization. These cases were previously referred to as “dementia without specific histological features,” but this term was used before the newer histologic stains for tau and ubiquitin proteins entered routine use. This nonspecific pattern of neuronal loss, gliosis, and microvacuolization was reported in 2 cases as a pathologic underpinning of PPA. [21] Similar changes have also been reported by Morris et al [24] under the term “hereditary dysphasic dementia” and by Neary, Snowden, and colleagues under the term frontal lobe dementia. [10]
As previously noted, most non-Alzheimer disease pathologies can be divided into those with positive staining for tau proteins, including those linked to chromosome 17, and frontotemporal lobar degeneration with ubiquitin staining (FTLD-U), due to progranulin or C9ORF72 mutations, leaving only rare cases with truly nonspecific pathology. [20]
Among the tau-positive patients, some develop symptoms and show pathologic criteria for corticobasal degeneration and others show overlap with the progressive supranuclear palsy (PSP) pathology.
Pharmacologic Therapy
Treatment of depression with a selective serotonin reuptake inhibitor (SSRI), such as paroxetine, sertraline, or citalopram, is frequently helpful. Trazodone may be helpful for sleep and for behavioral aberrations. These agents have been shown to be effective in small clinical trials. [14, 15, 16]
Neurotransmitter-based treatments, analogous to the use of dopaminergic agents in Parkinson disease or anticholinesterase agents in Alzheimer disease, have not proven beneficial in frontotemporal lobe dementia (FTD). There is not clearly a rationale for use of anticholinesterase drugs, such as donepezil (Aricept), rivastigmine (Exelon), or galantamine (Razadyne), since there is no definite cholinergic deficiency in FTD, [17] but these drugs are widely used. Anecdotally, they may improve memory but may worsen behavioral symptoms. The drug memantine has been thought to be helpful, but 2 recent, small clinical trials have not supported a benefit of this agent. [18, 19]
Investigational treatments
All of the pharmacologic treatments listed below must be considered investigational and not recommended for general use.
Dopaminergic drugs have been tested in patients with transcortical motor aphasia secondary to strokes. Anecdotal experience with dopamine agonist agents such as bromocriptine (Parlodel), pergolide (Permax), pramipexole (Mirapex), and ropinirole (Requip) has been unimpressive. Bromocriptine and pergolide are no longer available.
Stimulant drugs such as amphetamines and modafinil (Provigil) may benefit patients with frontal lobe syndromes. Large, randomized, double-blind studies are lacking. There is concern that the stimulant drugs might worsen behavior in patients with behavioral variant FTD.
As stated previously, the last few years have witnessed major breakthroughs in the understanding of the molecular genetics of these disorders, and we hope that treatment breakthroughs will follow.
Questions & Answers
Overview
What is frontotemporal dementia (FTD)?
What are the signs and symptoms of frontotemporal dementia (FTD)?
How is frontotemporal dementia (FTD) treated?
What causes frontotemporal dementia (FTD)?
What is the role of tau protein in the pathogenesis of frontotemporal dementia (FTD)?
What is the role of genetic mutations in the pathogenesis of frontotemporal dementia (FTD)?
What is the link between Alzheimer's disease and frontotemporal dementia (FTD)?
What is the link between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)?
What is the genetic distribution of frontotemporal dementia (FTD)?
What is the prevalence of frontotemporal dementia (FTD) in the US?
What is the global prevalence of frontotemporal dementia (FTD)?
Which patient groups are at highest risk for frontotemporal dementia (FTD)?
What is the prognosis of frontotemporal dementia (FTD)?
What is progressive nonfluent aphasia in frontotemporal dementia (FTD)?
What is semantic dementia in frontotemporal dementia (FTD)?
What is logopenic progressive aphasia in frontotemporal dementia (FTD)?
What are the changes in behavioral characteristic of frontotemporal dementia (FTD)?
What are the signs and symptoms of motor neuron disease in frontotemporal dementia (FTD)?
What are the signs and symptoms of less common frontotemporal dementia (FTD) variants?
Which physical findings are characteristic of frontotemporal dementia (FTD)?
Which conditions should be included in the differential diagnoses of frontotemporal dementia (FTD)?
What is the role of lab testing in the diagnosis of frontotemporal dementia (FTD)?
What are the diagnostic criteria for the behavioral variant of frontotemporal dementia (bvFTD)?
What is the role of imaging studies in the diagnosis of frontotemporal dementia (FTD)?
What is the role of SPECT and PET scanning in the diagnosis of frontotemporal dementia (FTD)?
What histologic findings are characteristic of frontotemporal dementia (FTD)?
What is the role of drug treatment for frontotemporal dementia (FTD)?
-
Hematoxylin and eosin stain of the left frontal cortex from a patient with primary progressive aphasia. This shows loss of neurons, plump astrocytes (arrow), and microvacuolation of the superficial cortical layers. Reproduced with permission of John Wiley & Sons, Inc.
-
Patient with progressive nonfluent aphasia. MRI showing focal, left temporal atrophy. Reprinted from Neurology in Clinical Practice, 4th ed. Kirshner H. Language and Speech Disorders. Copyright 2004, with permission from Elsevier.
-
Patient with progressive nonfluent aphasia. Positron emission tomography (PET) scan indicating hypometabolism of glucose in the left hemisphere. Reprinted from Neurology in Clinical Practice, 4th ed. Kirshner H. Language and Speech Disorders. Copyright 2004, with permission from Elsevier.
Tables

What would you like to print?
- Overview
- Etiology
- Genetic Distribution and Variation
- Epidemiology
- Prognosis
- Primary Progressive Aphasia
- Behavioral Changes
- Additional Presentations
- Physical Examination
- Differential Diagnosis
- Lab and EEG Studies
- Language and Neuropsychological Testing
- Imaging Studies
- Histologic Findings
- Pharmacologic Therapy
- Questions & Answers
- Show All
- Media Gallery
- References