Practice Essentials
Astrocytomas are a form of glioma (ie, a neoplasm of the glial cells, which constitute the supportive tissue of the brain and nervous system). The predominant cell type in these tumors is thought to be derived from an immortalized astrocyte. [1] Astrocytomas can arise anywhere in the nervous system, but most commonly occur in the brain. (See the image below.) Astrocytomas can be indolent or aggressive, depending on tumor grade, which drives prognosis and clinical decision making.
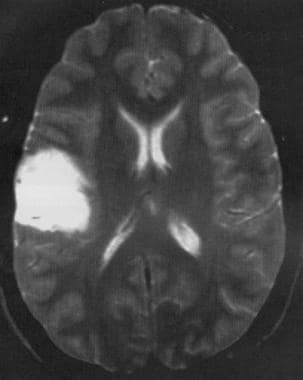 Axial T2-weighted MRI shows a low-grade astrocytoma of the inferior frontal lobe with mild mass effect and no surrounding edema.
Axial T2-weighted MRI shows a low-grade astrocytoma of the inferior frontal lobe with mild mass effect and no surrounding edema.
Signs and symptoms
Neurologic signs and symptoms from astrocytoma depend foremost on the site and extent of tumor growth in the CNS but may include any of the following:
-
Altered mental status
-
Cognitive impairment
-
Headaches
-
Nausea and vomiting
-
Visual disturbances
-
Motor impairment
-
Seizures
-
Sensory anomalies
-
Ataxia
Astrocytomas of the spinal cord or brainstem are less common and present as motor/sensory or cranial nerve deficits referable to the tumor's location.
On physical examination, patients may demonstrate localizing and lateralizing signs such as the following:
-
Cranial nerve palsies
-
Hemiparesis
-
Sensory levels
-
Alteration of deep tendon reflexes (DTRs)
-
Pathologic reflexes (eg, Hoffman sign, Babinski sign)
See Presentation for more detail.
Diagnosis
No laboratory studies are diagnostic of astrocytoma, but the following baseline laboratory studies may be obtained for general metabolic surveillance and preoperative assessment:
-
Basic metabolic profile
-
CBC
-
Prothrombin time (PT)
-
Activated partial thromboplastin time (aPTT)
MRI:
-
MRI is considered the standard imaging study.
-
Astrocytomas are generally isointense on T1-weighted images and hyperintense on T2-weighted images.
-
While low-grade astrocytomas (WHO grades 1-2) uncommonly enhance on MRI, many grade 3-4 astrocytomas enhance with gadolinium contrast agents.
-
High-resolution MRI is now often used to provide intraoperative image guidance.
CT:
-
A CT scan may be useful in the acute setting or when MRI is contraindicated.
-
On CT, low-grade astrocytomas appear as poorly defined, homogeneous, low-density masses without contrast enhancement; however, slight enhancement, calcification, and cystic changes may be evident.
-
High-grade astrocytomas may appear as low-density lesions or inhomogeneous lesions, with areas of both high and low density within the same lesion; unlike low-grade lesions, partial contrast enhancement is common.
-
Systemic imaging, generally consisting of a contrast-enhanced CT scan of the chest, abdomen, and pelvis, may be warranted to evaluate for the possibility of an alternate primary lesion.
Other studies:
-
Angiography may be used to rule out vascular malformations and to evaluate tumor blood supply
-
PET, SPECT, or technetium-based imaging can permit study of tumor metabolism and brain function
-
EEG may be used to evaluate and monitor epileptiform activity
-
ECG and chest radiographs are indicated to evaluate operative risk
Tissue diagnosis and WHO classification
Classification of astrocytomas is based on distinct histopathologic and molecular alterations, and drives treatment decision making. Therefore, tissue diagnosis (via biopsy or surgical resection) is often necessary prior to further treatment planning. This article focuses on the widely accepted updated World Health Organization (WHO) grading scheme, [2] and is confined to adult-type diffuse isocitrate dehydrogenase (IDH)-mutant gliomas, including grades 2-4 IDH1-mutant astrocytoma. See Background.
Management
Treatment options in astrocytomas include operative intervention, chemotherapy and radiotherapy, and are guided in part by WHO Classification. Therefore, tissue diagnosis with histological and molecular characterization is essential. Treatment decisions are best made by a team approach, including input from the involved neurosurgeon, radiation oncologist, and medical oncologist or neurologist, as well as the patient and/or their family. See Treatment and Medication.
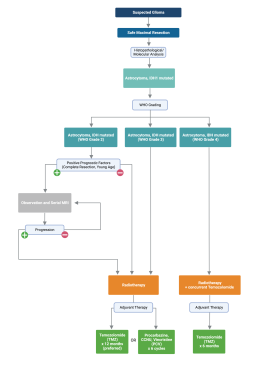
The multimodal treatment guidelines for IDH-mutated astrocytomas as recommended by the American Society of Clinical Oncology (ASCO) and Society of Neuro-Oncology (SNO) are summarized in the flow chart below:
Surgical care
-
Tumor resection is generally undertaken with the intent to cause the least possible neurologic injury to the patient.
-
Surgical resection provides improved survival advantage as well as histopathologic/molecular diagnosis of the tumor but is not curative. Total resection of an astrocytoma is often impossible because the tumors often exhibit tumor infiltration that is detectable only on a microscopic scale and/or invade eloquent regions of the brain.
-
New methods of tumor imaging are being developed to specifically fluorescently tag or label tumor cells so they may be visualized in the operating room, to guide surgical resection. Such methods used for astrocytomas include preoperative infusion of fluorescein or Gleolan (5-ALA/PPIX). [3]
-
Several imaging methods can be used preoperatively and intraoperatively to map functional areas of the brain, such as those that control speech, language, and motor and sensory functions, if the tumor abuts these regions, to ensure the safe resection of the lesion while avoiding eloquent brain structures. These methods include functional MRI (fMRI), diffusion tensor imaging (DTI) tractography, and intraoperative direct cortical/subcortical stimulation.
-
Stereotactic biopsy can be utilized to establish a tissue diagnosis in cases where surgical resection is not possible or preferred.
-
Diversion of CSF by external ventricular drain (EVD) or ventriculoperitoneal shunt (VPS) may be required to decrease intracranial pressure.
Radiation and Adjuvant chemotherapy
-
External beam radiotherapy is a mainstay of treatment for patients with IDH-mutant astrocytoma after surgical resection, but can be deferred in the setting of positive prognostic factors in grade 2 IDH-mutant astrocytomas (see Guidelines/Guidelines Summary).
-
Options for adjuvant chemotherapy include temozolomide (TMZ) as well as the combination of procarbazine, lomustine (CCNU), and vincristine (PCV), depending on the WHO grading (see Guidelines/Guidelines Summary).
Symptomatic therapy
-
Patients with an astrocytoma and a history of seizures should receive anticonvulsant therapy, with monitoring of the serum drug concentration; levetiracetam (Keppra) is often used.
-
Prophylactic use of anticonvulsants in astrocytoma patients with no prior history of seizures has been reported but remains controversial
-
The use of corticosteroids, such as dexamethasone, yields rapid improvement in most patients secondary to a reduction of tumor mass effect; patients receiving corticosteroids should have concurrent prophylaxis for gastrointestinal ulcers
For patient education information, see Brain Cancer.
Background
Astrocytomas constitute a broad group of gliomas, and have historically been classified on the basis of distinct radiographic and histologic features. Numerous grading schemes based on histopathologic characteristics have been devised, including the following:
-
Bailey and Cushing grading system
-
Kernohan grades I-IV
-
World Health Organization (WHO) grades 1-4
-
St. Anne/Mayo grades 1-4
Most recently, astrocytomas have been re-classified as part of the 2021 WHO Classification of Tumors of the Central Nervous System [2] (see the image below). This edition of the WHO grading scheme revolutionized classification by incorporating and emphasizing key molecular alterations, in addition to histopathologic features, in the diagnostic criteria for each tumor subtype.
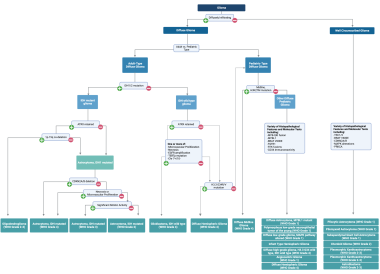
Broadly, two classes of astrocytic tumors are recognized: those with narrow zones of infiltration and well circumscribed (eg, pilocytic astrocytoma, subependymal giant cell astrocytoma, pleomorphic xanthoastrocytoma) and those with diffuse zones of infiltration (eg, low-grade astrocytoma, anaplastic astrocytoma, glioblastoma). For a discussion of well-circumscribed astrocytomas, see Pediatric Astrocytoma.
Diffuse gliomas share various features, including the following:
-
The ability to arise at any site in the CNS, with a preference for the cerebral hemispheres
-
Clinical presentation usually in adults
-
Heterogeneous histopathologic properties and biological behavior
-
Diffuse infiltration of contiguous and distant CNS structures, regardless of histologic stage
-
An intrinsic tendency to progress to more advanced grades
The updated WHO criteria also make a distinction between adult-type and pediatric-type diffuse gliomas. For a discussion of pediatric-type diffuse gliomas, see Pediatric Astrocytoma. The updated criteria emphasize the IDH mutational status as the primary molecular alteration used for stratification of adult-type diffuse gliomas, as mutations in either gene (IDH1 or IDH2) have been shown to alter disease progression, carry prognostic significance (the IDH1/2 mutation is associated with improved survival), and guide treatment decisions. The emphasis on this molecular marker, as demonstrated in the above flow chart, has led to two major changes in the classifications of astrocytoma:
-
Elimination of the entitity of IDH-mutant glioblastoma (now called WHO Grade 4 IDH-mutant astrocytoma)
-
Classification of any low-grade IDH-wildtype astrocytomas as primarily pediatric-type diffuse gliomas (in contrast, IDH-wildtype gliomas in adults tend to have one or more aggressive features, which allows them to be classified as glioblastoma WHO Grade 4; see Glioblastoma).
Oligodendroglioma, also IDH mutated, is distinguished from astrocytoma molecularly via existence of the chromosomal co-deletion 1p-19q and retention of the ATRX gene. For a detailed discussion of IDH-mutant oligodendroglioma, see Oligodendroglioma
Pathophysiology
Astrocytomas are a form of glial neoplasm that arises in the brain or spinal cord. Astrocytomas can be either well-circumscribed or diffusely infiltrating, which often drives the clinical manifestations, disease progression, treatment options, and prognosis.
Regional effects of astrocytomas include compression, invasion, and destruction of brain parenchyma. Arterial and venous hypoxia, competition for nutrients, release of metabolic end products (eg, free radicals, altered electrolytes, neurotransmitters), and release and recruitment of cellular mediators (eg, cytokines) disrupt normal parenchymal function. Elevated intracranial pressure (ICP) attributable to direct mass effect, increased blood volume, or increased cerebrospinal fluid (CSF) volume may mediate secondary clinical sequelae.
Neurologic signs and symptoms attributable to astrocytomas result from perturbation of CNS function. Focal neurologic deficits (eg, weakness, paralysis, sensory deficits, cranial nerve palsies) and seizures of various characteristics may permit localization of lesions. [4]
Infiltrating low-grade astrocytomas grow more slowly than their malignant counterparts. Several years often intervene between the initial symptoms and the establishment of a diagnosis of low-grade astrocytoma. Higher-grade astrocytomas tend to produce more-pronounced symptoms, focal neurologic symptoms, and functional impairment.
Etiology
The etiology of diffuse astrocytomas has been the subject of analytic epidemiological studies that have yielded associations with various disorders and exposures. [5] With the exception of therapeutic irradiation [6] and, perhaps, nitroso compounds (eg, nitrosourea), the identification of specific causal environmental exposures or agents has been unsuccessful. Although concern has been raised regarding cell phone use as a potential risk factor for development of gliomas, these claims are largely unsubstantiated. [7, 8, 9, 10, 11]
Children receiving prophylactic irradiation for acute lymphoblastic leukemia (ALL) have a 22-fold increased risk of developing CNS neoplasms, including WHO grade 2, 3, and 4 astrocytomas, with an interval for onset of 5-10 years. Furthermore, irradiation of pituitary adenomas has been demonstrated to carry a 16-fold increased risk of glioma formation. [12]
Evidence exists for genetic susceptibility to glioma development. For example, familial clustering of astrocytomas is well described in inherited neoplastic syndromes, such as Turcot syndrome, neurofibromatosis type 1 (NF1) syndrome, and p53 germ line mutations (eg, Li-Fraumeni syndrome). Biological investigation has found evidence that mutations in specific molecular pathways, such as the p53-MDM2-p21 and p16-p15-CDK4-CDK6-RB pathways, are associated with astrocytoma development and progression. Two-thirds of low-grade astrocytomas have p53 mutations. [13]
In patients with glioma who do not have single-gene alterations, anywhere between 5-10% have a family history of glial neoplasm, and having a first-degree relative with glioma doubles the risk of developing glioma. Large genome-wide association studies (GWAS) have identifed 25 risk loci associated with increased risk of glioma. Genetic loci specifically associated with IDH-mutant astrocytoma include portions of the IDH1 gene, among others. [14]
In addition, human leukocyte antigen (HLA) types have been associated with either increased or decreased risk for the development of brain gliomas. Machulla et al reported that, compared with a control population, patients positive for HLA-A*25 had a 3.0-fold increased risk of brain glioma (P = 0.04), patients positive for HLA-B*27, a 2.7-fold increased risk (P = 0.03), and patients positive for HLA-DRB1*15 had a 2.2-fold risk (P= 0.03), while those with HLA-DRB1*07 had a 0.4-fold decreased risk (P = 0.02). [15]
Epidemiology
The American Cancer Society estimates that in 2023, approximately 24,810 malignant tumors of the brain or spinal cord will be diagnosed, and about 18,990 deaths will occur from those tumors. [16] Brain and other nervous system tumors are the second leading cause of cancer death among males younger than 40 years old and females younger than 20 years old. [17]
The annual incidence of glioma in the United States is 6.0 cases per 100,000 population. [8] The majority of these cases (61.5%) were classified as glioblastomas (according to 2016 WHO criteria), while 18.8% were non-glioblastoma astrocytomas and the remainder were non-astrocytoma gliomas. [8] In an analysis of the Central Brain Tumor Registry of the United States (CBTRUS) from 2019, 19% of all adult-type diffuse gliomas were IDH1-mutant astrocytomas. [14]
Age
While increasing age has been shown to be a very strong risk factor for the development of glioblastoma, IDH1-mutant gliomas tend to affect a younger population. The median age at diagnosis of IDH1-mutant gliomas has been reported to be around 36-38 years (36 years for grades 2-3, 38 years for grade 4), while the median age for IDH1-wildtype gliomas (including glioblastomas) is around 50-60 years. [14]
Race
In the United States, the incidence of adult-type diffuse glioma is the highest in the non-Hispanic White population. Non-hispanic Whites have a 30% higher risk of glioma than Hispanic Whites, and double the risk of Black, Asian or Pacific Islander, and American Indian or Alaskan Native individuals. [14] However, demographic and socioeconomic factors, including health disparities and access to care, have been reported to influence measured distributions of disease incidence.
Sex
No clear sex predominance has been identified in the development of pilocytic astrocytomas. A slight male predominance, with a male-to-female ratio of 1.18:1, has been reported for development of lower-grade astrocytomas. A more significant male predominance, with a male-to-female ratio of 1.87:1, has been reported for the development of higher-grade astrocytomas. Five-year survival in non-glioblastoma astrocytoma is lowest in non-Hispanic Whites (44.1%) as compared with all other groups (50.8%). [14]
Prognosis
IDH-mutant astrocytomas are considered incurable, although treatment can prolong survival. However, prognosis for patients with IDH-mutant astrocytomas is significantly better than those with tumors without the IDH1 mutation, which is why molecular classification of the tumor (specifically IDH status) is critical for decision making and patient education.
While prognostic data are limited for tumors classified using the 2021 WHO criteria, median survival after diagnosis of IDH-mutant astrocytomas by grade is as follows [18, 19, 20, 21] :
-
WHO Grade 2: 10-12 years
-
WHO Grade 3: 8-10 years
-
WHO Grade 4: 3-4 years
Given the impact of the IDH mutation on prognosis, many attempts have been made to further elucidate prognosis and response to various treatment modalities based on tumor molecular profiling. For example, a homozygous deletion of the cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) gene portends a poor prognosis, and therefore automatically results in a WHO grade 4 classification in the 2021 criteria, independent of histopathologic criteria.
Beyond WHO grade, the following factors, among others, have been reported to have a negative impact on survival of patients with astrocytomas [22, 23] :
-
Male sex
-
Worse functional status
-
Larger tumors (> 5 cm)
Survivors of pediatric astrocytoma remain at high risk for long-term complications of their disease and its treatment. These patients require lifelong monitoring for late effects. [24]
-
Low-grade diffuse astrocytoma and low cellularity with minimal nuclear atypia.
-
Diffuse astrocytoma with microcyst formation.
-
Gemistocytic astrocytoma tumor cells have eosinophilic cytoplasm with nuclei displaced to the periphery.
-
Characteristic pilocytic astrocytoma, long bipolar tumor cells, and Rosenthal fibers.
-
Anaplastic astrocytoma with high cellularity with marked nuclear atypia.
-
Gross specimen of a low-grade astrocytoma.
-
Axial CT scan, precontrast and postcontrast, shows a low-grade astrocytoma of the left frontal lobe. The tumor is nonenhancing.
-
Coronal postcontrast T1-weighted MRI shows a low-grade astrocytoma in the right inferior frontal lobe just above the sylvian fissure. No enhancement is present post–gadolinium administration.
-
Axial T2-weighted MRI shows a low-grade astrocytoma of the inferior frontal lobe with mild mass effect and no surrounding edema.
-
IDH-mutant astrocytoma treatment guidelines.
-
Higher-grade IDH-mutant astrocytoma, with nuclear atypia and mitoses. Courtesy of Wikipedia (https://en.wikipedia.org/wiki/Anaplastic_astrocytoma).
-
2021 WHO classification of gliomas.









