Practice Essentials
Polycystic kidney disease is an inherited disease that involves bilateral kidney cysts. The condition is broadly divided into 2 forms: autosomal dominant polycystic kidney disease (ADPKD; see the image below) and autosomal recessive polycystic kidney disease (ARPKD). This article focuses on ADPKD; for full discussion of ARPKD, see Pediatric Polycystic Kidney Disease. However, note that although ADPKD was previously known as adult polycystic kidney disease and ARPKD was previously known as infantile polycystic kidney disease, those descriptions are not accurate, and that nomenclature is no longer used.
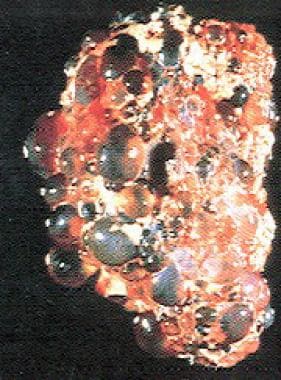
ADPKD is the most frequent genetic cause of chronic kidney disease (CKD) in adults, accounting for 6-10 % of patients on dialysis in the United States. [1] It is a multisystemic and progressive disorder characterized by cyst formation and enlargement in the kidney and other organs (eg, liver, pancreas, spleen). Clinical manifestations usually begin in the third to fourth decade of life, but cysts may be detectable in childhood and in utero. Up to 50% of patients with ADPKD require kidney replacement therapy (KRT) by 60 years of age. [2]
ARPKD is characterized by cystic dilatation of kidney collecting ducts, along with hepatic abnormalities of varying degrees, including biliary dysgenesis and periportal fibrosis. The disorder is usually diagnosed in infants and children, although hepatic involvement may not manifest in neonates (50-60%).
Just as ADPKD may involve the liver, autosomal dominant polycystic liver disease (ADPLD) may involve cysts in the kidneys, although if present, they are few in number. However, like patients with ADPKD, patients with ADPLP also present with abdominal pain, as the liver cysts enlarge and cause hepatomegaly.
Signs and symptoms
ADPKD is a multisystem disorder. Multiple kidney and extrarenal manifestations have been described that cause significant complications.
Pain—in the abdomen, flank, or back—is the most common initial complaint, and it is almost universally present in patients with ADPKD. Dull aching and an uncomfortable sensation of heaviness may result from a large polycystic liver.
The pain can be caused by any of the following:
-
Enlargement of one or more kidney cysts
-
Bleeding: May be confined inside the cyst or lead to gross hematuria with passage of clots or a perinephric hematoma
-
Urinary tract infection (UTI) (eg, acute pyelonephritis, infected cysts, perinephric abscess)
-
Nephrolithiasis and renal colic
-
Rarely, a coincidental hypernephroma
See Presentation for more detail.
Diagnosis
Examination in patients with ADPKD may demonstrate the following:
-
Hypertension: One of the most common early manifestations of ADPKD, and associated with rapid chronic disease progression [3]
-
Palpable, bilateral flank masses, in advanced ADPKD
-
Nodular hepatomegaly, in patients with severe polycystic liver disease
-
Rarely, symptoms related to advanced CKD (eg, pallor, uremic fetor, dry skin, edema)
Testing
Routine laboratory studies include the following:
-
Serum chemistry profile, including calcium and phosphorus
-
Cell blood cell count
-
Measurement of blood lipid concentrations
-
Urinalysis
-
Urine culture
-
Uric acid determination
-
Intact parathyroid hormone assay and vitamin D assay
Genetic testing can be performed when a precise diagnosis is needed and the results of imaging testing are indeterminate. For example, genetic testing is indicated in individuals at risk for ADPKD who are being considered as potential kidney donors, and for screening embryos in preimplantation genetic diagnosis. [4, 5]
Staging
Staging of ADPKD follows that of CKD and is based on the estimated glomerular filtration rate (GFR), as follows:
-
Stage 1: GFR > 90 mL/min
-
Stage 2: GFR 60-90 mL/min
-
Stage 3: GFR 30-60 mL/min
-
Stage 4: GFR 15-29 mL/min
-
Stage 5: GFR < 15 mL/min
Imaging studies
Radiologic studies used in the evaluation of ADPKD include the following:
-
Ultrasonography: Technique of choice for patients with ADPKD and for screening patients' family members; useful for exploring abdominal extrarenal features of ADPKD (eg, liver cysts, pancreatic cysts)
-
CT scanning: Not routine; used in complicated cases (eg, kidney stone, suspected tumor)
-
MRI: Not routine; helpful in distinguishing renal cell carcinoma from simple cysts; criterion standard to help determine total kidney volume for clinical trials of drugs for ADPKD
-
Magnetic resonance angiography (MRA): Not routine; preferred imaging technique for diagnosing ADPKD-related intracranial aneurysms
Ultrasound diagnostic criteria for ADPKD, developed by Ravine et al, are very useful for identifying patients at risk of pathogenic variants in PKD1. [6] The presence of fewer than 2 renal cysts has a negative predictive value of 100% and can be considered sufficient to exclude the disease in at-risk individuals over 40 years of age. The diagnosis of ADPKD is established by any of the following:
-
At least 2 kidney cysts or 1 cyst in each kidney in patients younger than 30 years
-
At least 2 cysts in each kidney in patients aged 30-59 years old.
-
At least 4 cysts in each kidney in patients aged 60 years or older
As the Ravine criteria are less sensitive for individuals with mutations in PKD2, Pei et al proposed the following ultrasonographic diagnostic criteria for ADPKD due to either PKD1 or PKD2 mutations, to screen patients with a family history of ADPKD but unknown genotype [4] :
-
Three or more kidney cysts (unilateral or bilateral) in patients aged 15 to 39 years
-
Two or more cysts in each kidney in patients aged 40 to 59 years
-
Four or more cysts in each kidney in patients aged ≥60 years
-
Fewer than two renal cysts in at-risk individuals aged ≥40 years excludes the disease
Indications for MRA are as follows [7, 5] :
-
Family history of stroke, intracranial aneurysm (ICA), or hemorrhage; patients with a family history of ICA and a negative screening study should be rescreened at 5-10–year intervals
-
Before major elective surgery
-
Central nervous system signs or symptoms (eg, nausea and vomiting, lethargy, photophobia, focal signs, seizure, transient ischemic attack, loss of consciousness)
-
High-risk occupation or hobby, in which a loss of consciousness may be lethal (eg, airline pilot)
-
New-onset severe headache
-
Patient anxiety despite adequate information
See Workup for more detail.
Management
Management of ADPKD includes the following:
-
Control blood pressure: Drugs of choice are ACEIs (eg, enalapril, lisinopril) or ARBs (eg, valsartan, telmisartan, losartan, irbesartan, candesartan, olmesartan)
-
Control abnormalities related to advanced CKD: Drugs to maintain electrolyte levels (eg, calcium carbonate, calcium acetate, sevelamer, lanthanum carbonate, calcitriol, diuretics)
-
Treat kidney and liver cyst infections: Gyrase inhibitors (eg, ciprofloxacin, ceftriaxone, clindamycin); dihydrofolic acid inhibitors (TMX/SMP)
-
Treat hematuria: Copious oral hydration; consider analgesics
-
Reduce abdominal pain caused by enlarged kidneys
-
Prevent cardiac valve infection in patients with intrinsic valve disease
-
Slow kidney function decline in adults at risk of rapidly progressive ADPKD (tolvaptan)
Surgical intervention in ADPKD includes the following:
-
Surgical drainage: Usually in conjunction with ultrasonography- or CT-guided puncture; in cases of infected kidney/liver cysts not responding to conventional antibiotics
-
Open or fiberoptic-guided surgery: For excision/drainage of the outer walls of cysts to relieve symptoms
-
Nephrectomy: Last resort for control of pain or hematuria in patients with inaccessible cysts in the renal medullae; bilateral nephrectomy in patients with severe hepatic involvement
-
Partial hepatectomy: To manage massive hepatomegaly
-
Liver transplantation: In very rare cases of portal hypertension due to polycystic liver or hepatomegaly with nonresectable areas
Patients with ADPKD who progress to KRT may require the following procedures:
-
Hemodialysis
-
Peritoneal dialysis
-
Kidney transplantation
See Treatment and Medication for more detail.
Pathophysiology
The main feature of ADPKD is a bilateral progressive increase in the number of cysts, which may reduce kidney function to the point where the individual requires kidney replacement therpy (KRT). Hepatic cysts, intracraneal aneurysms, and cardiac valvular abnormalities also may occur.
Although ADPKD is a systemic disease, it shows a focal expression; less than 1% of nephrons become cystic. In ADPKD, each epithelial cell within a renal tubule harbors a germ-line mutation, yet only a tiny fraction of the tubules develop kidney cysts.
It is currently held that the cells are protected by the allele inherited from the parent without ADPKD. When this allele is inactivated by a somatic event (eg, mutation) within a solitary renal tubule cell, the cell divides repeatedly until a cyst develops, with an aberrant growth program causing unchecked expansion. [8] The severity of ADPKD is thought to be a direct consequence of the number of times and the frequency with which this cystogenic process occurs within the kidneys over the life of the patient.
The hyperplastic cells cause an out-pocketing of the tubule wall, with the formation of a saccular cyst that fills with fluid derived from glomerular filtrate that enters from the afferent tubule segment. Progressive expansion eventually causes most of the emerging cysts to separate from the parent tubule, leaving an isolated sac that fills with fluid by transepithelial secretion. This isolated cyst expands relentlessly as a result of continued proliferation of the mural epithelium together with the transepithelial secretion of sodium chloride and water into the lumen. [8]
The expanding fluid-filled tumor masses elicit secondary and tertiary changes within the renal interstitium evinced by thickening and lamination of the tubule basement membranes, infiltration of macrophages, and neovascularization. Fibrosis within the interstitium begins early in the course of the disease.
Cellular proliferation and fluid secretion may be accelerated by cyclic adenosine monophosphate (cAMP) and growth factors such as epidermal growth factor (EGF). In summary, cysts function as autonomous structures and are responsible for progressive kidney enlargement in ADPKD. [2, 9]
Approximately 85-90% of patients with ADPKD have an abnormality in the PKD1 gene located on the short arm of chromosome 16. Most of the remaining 10-15% of ADPKD cases are caused by pathogenic variants in the PKD2 gene, which is located on the long arm of chromosome 4. A third candidate gene, GANAB (glucosidase II alpha subunit), accounting for a very low number of ADPKD cases, has also been described. [10] Additional cases caused by mutations in ALG9, DNAJB11, or LRP5 have been reported. [11, 12, 13]
PKD1 and PKD2 are expressed in most organs and tissues of the human body. The proteins that are encoded by PKD1 and PKD2, polycystin 1 (PC1) and polycystin 2 (PC2), seem to function together to regulate the morphologic configuration of epithelial cells. The polycystins are expressed in development as early as the blastocyst stage and are expressed in a broad array of terminally differentiated tissues. PC1 and PC2 belong to a subfamily of transient receptor potential (TRP) channels. The functions of the polycystins have been scrutinized to the greatest extent in epithelial tissues of the kidneys and liver and in vascular smooth muscle. [8, 14] (See Etiology.)
A decrease in urine-concentrating ability is an early manifestation of ADPKD. Plasma vasopressin levels are increased; this may represent the body's attempt to compensate for the reduced concentrating capacity of the kidneys and could contribute to the development of renal cysts, hypertension, and kidney insufficiency. [15]
Bleeding
Renal cysts in ADPKD are associated with excessive angiogenesis evinced by fragile vessels stretched across their distended walls. When traumatized, these vessels may leak blood into the cyst, causing it to expand rapidly, resulting in excruciating pain. If bleeding continues, then the cyst may rupture into the collecting system, causing gross hematuria. Alternatively, the cyst may rupture into the subcapsular compartment and eventually dissect through the renal capsule to fill the retroperitoneal space.
Etiology
ADPKD is a hereditary disorder with an autosomal dominant pattern of inheritance. The disorder occurs equally in males and females. Each offspring of an affected person has a 50% chance of inheriting the genetic variant responsible for the disease.
ADPKD is a genetically heterogeneous condition that involves at least 2 genes. PKD1 is located on chromosome 16p13.3 and accounts for most ADPKD cases. PKD2 is located on chromosome 4q21-q22 and accounts for up to 15% of ADPKD cases. Other genes identified as rare causes of ADPKD include GANAB, ALG9, DNAJB11, and LRP5. [11, 12]
Polycystin 1 and 2
PKD1 codes for a 4304–amino acid protein, polycystin 1 (PC1). The function of PC1 is not yet fully defined, but this protein interacts with polycystin 2 (PC2) and is involved in cell cycle regulation and intracellular calcium transport. PC1 localizes in the primary cilia of renal epithelial cells, which function as mechanosensors and chemosensors.
PKD2 codes for PC2, a 968–amino acid protein that is structurally similar to PC1 and co-localizes to the primary cilia of renal epithelial cells. It is a member of the family of voltage-activated calcium channels.
PC1 and PC 2 are highly conserved, ubiquitous transmembrane proteins. In the kidney, they are located in the epithelial cells of the renal tubules—in particular, in the primary cilia at the luminal side of the tubules, as well as in other areas of the renal cell epithelium.
PC1 is a large protein with a long extracellular N-terminal region, 11 transmembrane domains, and a short intracellular C-terminal tail. PC2 is structurally related to the transient receptor potential (TRP) channel family, and it is known to function as a nonselective cation channel permeable to Ca2+.
PC1 and PC2 form heteromeric complexes and co-localize in the primary cilium of renal epithelial cells. The primary cilium is a long, nonmotile tubular structure located in the apical surface of the epithelial cells in the renal tubules. Its function was unknown for a long time, but studies now indicate that the primary cilium may be a mechanoreceptor that senses changes in apical fluid flow and transduces them into an intracellular Ca2+ signaling response [9] .
This model involves the participation of PC as a mechanical sensor of ciliary bending induced by luminal fluid flow. Bending of the cilium would cause a conformational change in PC1 that would, in turn, activate the PC2–associated Ca2+ channel, increasing the intracellular Ca2+ concentration and triggering intracellular signaling pathways leading to normal kidney development. [16]
There is a genotype-phenotype correlation for PKD1 mutations. Truncating mutations cause a more severe phenotype than non-truncating ones. [17]
In general, patients with PKD1 pathogenic variants present with more severe disease than patients with PKD2 pathogenic variants. The mean age of requiring kidney replacement therapy is 53 years in patients with PKD1 pathogenic variants, but is 74 years in patients with PKD2 pathogenic variants. [5]
The genetic heterogeneity of ADKPD, and the possible contribution of modifier genes, may explain the wide clinical variability in this disease, both within and among families. [13]
Epidemiology
Worldwide, ADPKD affects approximately 4 to 7 million individuals and accounts for 7-15% of patients on kidney replacement therapy (KRT). In North America and Europe, ADPKD is responsible for 6-10% of KRT cases. [18, 2] Approximately one per 800-1000 population carries a pathogenic variant for this condition. Approximately 85-90% of those individuals have PKD1 pathogenic variants; most of the remainder have PKD2 disease-causing variants.
ADPKD is slightly more severe in males than in females. [19]
Symptoms generally increase with age. Children very rarely present with advanced chronic kidney disease from ADPKD.
Prognosis
The prognosis in patients with ADPKD covers a wide spectrum. Typically, however, ADPKD causes progressive kidney dysfunction, resulting in grossly enlarged kidneys and kidney failure by the fourth to sixth decade of life. There is an inverse association between the size of polycystic kidneys and the glomerular filtration rate (GFR). [2]
By the time kidney function begins to decline, the kidneys are usually markedly enlarged and distorted, with little visible parenchyma on imaging studies. At this stage, the average rate of estimated GFR decline is 4.4 to 5.9 mL/min per year. Up to 77% of patients are alive with preserved kidney function at age 50 years, and 52% at age 73 years. Men tend to progress to advanced chronic kidney disease more rapidly and require kidney replacement therapy (KRT) at a younger age than do women.
The presence of more than one risk factor increases the risk of progression to KRT. [1] Risk factors for progression include the following:
-
PKD1 genotype
-
Kidney size
-
First episode of hematuria before age 35 years
-
Severe and frequent kidney infections
-
Hypertension onset before age 35 years
-
Multiple pregnancies
-
Black racial background
-
Male sex
Although PKD1 and PKD2 pathogenic variants result in ADPKD with similar clinical features, they impart strikingly different kidney prognoses. [20] Patients with PKD2 pathogenic variants show a milder disease: their median age for initiation of KRT is 68 years, compared with 56 years in individuals with PKD1 variants. Nevertheless, even though PKD2 phenotype is milder than PKD1, it has an overall impact on survival and shortens life expectancy. [17] .
Cardiovascular pathology and infections account for approximately 90% of deaths in patients treated with hemodialysis or peritoneal dialysis and after kidney transplantation. A rare cause of mortality is in ADPKD is subarachnoid hemorrhage from intracranial aneurysms.
The Mayo Clinic calculator for ADPKD is a useful tool for predicting disease progression. Recommendations for assessing rapid progression of ADPKD have been provided by European experts. [5, 21, 22] The French PROPKD score predicts risk of progression to KRT in patients with ADPKD. The score is calculated on the basis of the following factors [19] :
-
Male sex: 1 point
-
Hypertension before 35 years of age: 2 points
-
First urologic event before 35 years of age: 2 points
-
PKD2 pathogenic variant: 0 points
-
Nontruncating PKD1 pathogenic variant: 2 points
-
Truncating PKD1 pathogenic variant: 4 points
Risk categories, on the basis of point totals, are as follows:
-
0-3 points: Low risk; median age for KRT onset 70.6 years
-
4-6 points: Intermediate risk; median age for KRT onset 56.9 years
-
7-9 points: High risk; median age for KRT onset 49 years
A PROPKD score of 3 or less eliminates evolution to KRT before 60 years of age, with a negative predictive value of 81.4%. A score higher than 6 forecasts KRT onset before 60 years of age, with a positive predictive value of 90.9%. [19]
Patient Education
Ensure that patients are aware that this disease is hereditary and that their children have a 50% chance of acquiring the disease. Patients should also understand that although several treatments are being tested, this disease currently has no cure. Only interventions that slow the progression of kidney disease (eg, adequate blood pressure control, tolvaptan treatment) are of benefit. Hopefully, effective specific therapy will be available in a few years.
Prenatal diagnosis is available through genetic testing. Suggest that family members who are not screened for ADPKD have annual blood pressure checks and ultrasound screenings for kidney cyst diagnosis.
Patient education information is available at Living With Autosomal Dominant Polycystic Kidney Disease.
-
Polycystic kidney.
-
Polycystic kidney disease and massive polycystic liver disease.






