Practice Essentials
Primary myelofibrosis is a clonal disorder arising from the neoplastic transformation of early hematopoietic stem cells. [1, 2, 3, 4] Older terms for this disorder include agnogenic myeloid metaplasia with myelofibrosis and chronic idiopathic myelofibrosis. [5] Primary myelofibrosis is categorized as a chronic myeloproliferative disorder, along with chronic myelogenous leukemia (CML), polycythemia vera, and essential thrombocytosis. (See Etiology.) [6]
The disorder is characterized by the following (see Workup):
-
Anemia
-
Bone marrow fibrosis (myelofibrosis)
-
Extramedullary hematopoiesis
-
Leukoerythroblastosis and teardrop-shaped red blood cells (RBCs) in peripheral blood (see the image below)
-
Hepatosplenomegaly
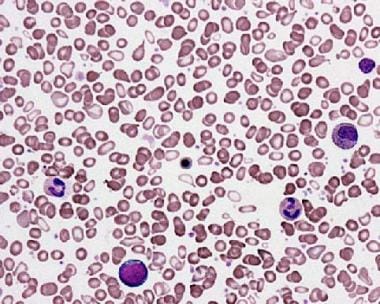 Primary myelofibrosis. Peripheral smear shows teardrop red blood cells (RBCs) and a leukoerythroblastic picture with nucleated RBC precursors and immature myeloid cells. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.
Primary myelofibrosis. Peripheral smear shows teardrop red blood cells (RBCs) and a leukoerythroblastic picture with nucleated RBC precursors and immature myeloid cells. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.
Treatment for primary myelofibrosis is tailored to disease severity, as follows:
-
Observation alone may be appropriate for low-risk, asymptomatic disease.
-
Mild cases may require only supportive therapy.
-
Higher-risk disease may respond to treatment with a JAK inhibitor.
-
Refractory splenomegaly may be an indication for splenectomy.
-
Allogeneic stem cell transplantation is potentially curative and should be considered for patients at high risk.
See Treatment and Medication.
Complications
Portal hypertension occurs in approximately 7% of patients with primary myelofibrosis and may be related to increased portal flow resulting from marked splenomegaly and to intrahepatic obstruction resulting from thrombotic obliteration of small portal veins. This may result in variceal bleeding or ascites. Hepatic or portal vein thrombosis may occur. Symptomatic portal hypertension is managed by splenectomy, with or without the creation of a portosystemic shunt. (See Presentation, Workup, and Treatment.)
Splenic infarction may occur and results in an acute or subacute onset of severe pain in the left upper quadrant that may be associated with nausea, fever, and referred left shoulder discomfort. The episode is usually self-limited and may last several days. Treat patients with hydration and opioid analgesics. Individuals with refractory cases of primary myelofibrosis may require splenectomy or splenic irradiation. (See Presentation and Treatment.)
Extramedullary hematopoiesis may involve any organ; symptoms depend on the organ or site of involvement. It may result in gastrointestinal (GI) tract bleeding, spinal cord compression, seizures, hemoptysis, and/or effusions. These are easily controlled with low-dose radiation. (See Treatment.)
Patients with primary myelofibrosis are also prone to developing infectious complications because of defects in humoral immunity.
Osteosclerosis, hypertrophic osteoarthropathy, and periostitis may occur, resulting in significant pain and discomfort. This may require the administration of nonsteroidal anti-inflammatory drugs (NSAIDs) or opioid analgesics. Gout or urate stones may develop as a result of uric acid overproduction. Allopurinol should be used to keep the serum uric acid level in the reference range.
For discussion of myelofibrosis in children, see Pediatric Myelofibrosis. For patient education information, see the Blood Cancer Directory, as well as Anemia.
Pathophysiology
In patients with primary myelofibrosis, the hematopoietic system is most affected. Other organ systems may be involved via extramedullary hematopoiesis. (See the image below.)
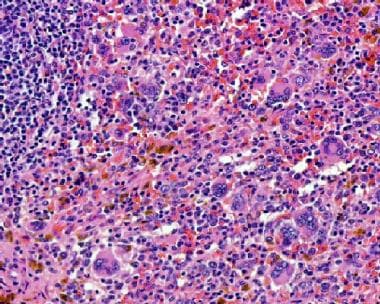 Extramedullary hematopoiesis in the spleen of a patient with primary myelofibrosis. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.
Extramedullary hematopoiesis in the spleen of a patient with primary myelofibrosis. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.
Clonality studies in patients with primary myelofibrosis demonstrate that myeloid cells arise from clonal stem cells; however, bone marrow fibroblasts and, sometimes, T cells are polyclonal. The cause of the excessive marrow fibrosis observed in primary myelofibrosis remains unclear.
Platelets, megakaryocytes, and monocytes are thought to secrete several cytokines, such as transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), interleukin 1 (IL-1), epidermal growth factor (EGF), and basic fibroblast growth factor (bFGF), which may result in fibroblast formation and extracellular matrix proliferation. In addition, endothelial proliferation and growth of capillary blood vessels in the bone marrow are observed and may be a result of TGF-β and bFGF production.
Neoangiogenesis is a hallmark feature of chronic myeloproliferative disorders. Approximately 70% of patients with primary myelofibrosis have substantial increases in bone marrow microvessel density. Neoangiogenesis in primary myelofibrosis is noted in medullary and extramedullary hematopoiesis. Increased serum vascular endothelial growth factor levels have been postulated as the underlying mechanism for increased angiogenesis.
Etiology
Approximately 50-60% of patients with primary myelofibrosis have a gain-of-function mutation in the Janus kinase 2 (JAK2) gene, the JAK2 V617F mutation, which leads to increased cytokine responsiveness of myeloid cells. Another 5-10% of patients have somatic mutations of JAK2 exon 12 or activating mutations of the thrombopoietin receptor gene MPL. In two separate studies, Klampfl et al and Nangalia et al found that mutations in the gene encoding calreticulin (CALR) were present in the majority of patients who lacked mutations in JAK2 or MPL. [7, 8] Mutations in NRAS, KRAS, PTPN11, GATA2, TP53, and RUNX1 have been found in < 5% of patients. [9]
No specific risk factors can be identified in most patients with primary myelofibrosis. However, exposure to radiation, Thorotrast contrast agents, and industrial solvents (eg, benzene, toluene) have been associated with increased risk. [10, 11, 12, 13]
Epidemiology
Primary myelofibrosis is an uncommon disease, with an annual incidence of approximately 0.5-1.5 cases per 100,000 individuals in the United States. In a review of European data sources, the incidence rate of myelofibrosis ranged from 0.1 to 1 cases per 100,000 per year. [14]
Race-, sex-, and age-related demographics
Primary myelofibrosis appears to be more common in Whites than in individuals of other races. In addition, an increased prevalence rate of the disorder has been noted in Ashkenazi Jews.
A slight male preponderance appears to exist for primary myelofibrosis; however, in younger children, girls are affected twice as frequently as boys.
Primary myelofibrosis characteristically occurs in individuals over age 50 years, with the median age at diagnosis being approximately 65 years. However, the disease has been reported in persons in all phases of life, from neonates to octogenarians.
Approximately 22% of affected patients are younger than 56 years. Primary myelofibrosis in children usually occurs in the first 3 years of life.
Prognosis
The median length of survival for patients with primary myelofibrosis is 3.5-5.5 years. The 5-year survival rate is about half of that expected for age- and sex-matched controls. Fewer than 20% of patients are expected to be alive at 10 years. [15] The common causes of death in patients with primary myelofibrosis are infections, hemorrhage, cardiac failure, postsplenectomy mortality, and transformation into acute leukemia. Leukemic transformation occurs in approximately 20% of patients with primary myelofibrosis within the first 10 years.
Advanced age and anemia are associated with shorter survival. Renal failure, hepatic failure, and thrombosis have also been reported as causes of death.
Other poor prognostic factors include the following:
-
Hypercatabolic symptoms
-
Leukocytosis (leukocyte count of 10,000-30,000/μL)
-
Leukopenia
-
Circulating blasts
-
Increased numbers of granulocyte precursors
-
Thrombocytopenia (platelet count of < 100,000/μL)
-
Karyotype abnormalities
Bone marrow vascularity is significantly increased in patients with primary myelofibrosis. Increased bone marrow microvascular density has also been reported in approximately 70% of patients with primary myelofibrosis, and it is an independent poor prognostic factor for survival.
A study of 570 patients with primary myelofibrosis found that prognostic significance exists for carriage of mutations in CALR (favorable) and ASXL1 (unfavorable). Patients who were CALR + and ASXL1- had the longest survival (median 10.4 years), whereas those who were CALR- and ASXL1+ had the shortest survival (median 2.3 years). Patients who were either CALR+ and ASXL1+ or CALR- and ASXL1- had similar rates of survival (median 5.8 years). The prognostic model was independent of the Dynamic International Prognostic Scoring System (DIPSS; see below). [16]
Rozovski and colleagues developed a prognostic model for primary myelofibrosis consisting of four elements: age and mutations in the Janus kinase 2 (JAK2), CALR, and myeloproliferative leukemia virus (MPL) genes. By itself, the JAK2V617F allele burden proved to have prognostic significance: median overall survival (OS) was 80 months in patients with a JAK2V617F allele burden of 50% or over, versus 50 months in those with a JAK2V617F allele burden of less than 50% (P=0.01). [17]
The best prognosis, with a median survival of 126 months, was seen in patients aged 65 years or under who had a low JAK2V617F allele burden and CALR and MPL mutations. The worst was in patients older than 65 years with a low JAK2V617F allele burden or no JAK2, CALR, or MPL mutations (“triple negative”) who had a median survival of only 35 months. Intermediate survival duration occurred in patients with one risk factor. [17]
Scoring systems
Several scoring systems to determine the prognosis in primary myelofibrosis have been developed.
DIPSS-plus
The Dynamic International Prognostic Scoring System–plus (DIPSS-plus) for primary myelofibrosis uses the following eight adverse factors to predict survival [18] :
-
Age older than 65 years
-
Hemoglobin level lower than 10 g/dL
-
Leukocyte count higher than 25 × 10 9/L
-
Platelet count lower than 100 × 10 9 /L
-
Circulating blasts of 1% or more
-
Constitutional symptoms
-
Red blood cell transfusion dependency
-
Unfavorable karyotype (ie, complex karyotype or sole or two abnormalities that include +8, -7/7q-, i(17q), inv(3), 5/5q-, 12p-, or 11q23 rearrangement)
DIPSS-plus classifications and median survival times are as follows:
-
Low risk (0 adverse points): 15.4 years
-
Intermediate-1 risk (1 adverse point): 6.5 years
-
Intermediate-2 risk (2-3 adverse points): 2.9 years
-
High risk (4-6 adverse points): 1.3 years
MIPSS70
The MIPSS70 (mutation-enhanced international prognostic scoring system for transplant-age patients) includes clinical risk variables and mutations; subsequent versions include karyotype (MIPSS70+ and MIPSS70+ version 2.0). This system was developed for transplant decision-making in patients age 70 years or younger. An online score calculator is available for MIPSS70 and MIPSS70+ version 2.0. [19]
MIPSS70 is based on three genetic variables and six clinical risk factors. The genetic variables are as follows:
-
Absence of CALR type 1/like mutation
-
Presence of any high molecular risk [HMR] mutation, specifically ASXL1, SRSF2, EZH2, IDH1, or IDH2
-
Presence of ≥2 HMR mutations)
The clinical risk factors are as follows:
-
Hemoglobin < 10 g/dL
-
Leukocytes >25 × 10 9/L
-
Platelets < 100 × 10 9/L
-
Circulating blasts ≥2%
-
Bone marrow fibrosis grade ≥2
-
Constitutional symptoms
MIPSS70+ added “unfavorable” karyotype as a fourth genetic variable, and reduced the number of clinical risk factors to four (hemoglobin < 10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥2%, and constitutional symptoms). Those clinical and genetic risk factors were used to classify three risk categories for MIPSS70 (low, intermediate, and high) and four risk categories for MIPSS70+ (low, intermediate, high, and very high).
In the most recent version, MIPSS70+ 2.0, clinical risk variables and points are as follows:
-
Severe anemia (hemoglobin < 8 g/dL in women, < 9 g/dL in men): 2 points
-
Moderate anemia (hemoglobin 8–9.9 g/dL in women, 9–10.9 g/dL in men): 1 point
-
Circulating blasts ≥2%: 1 point
-
Constitutional symptoms: 2 points
Genetic variables and points are as follows::
-
Very high risk (VHR) karyotype (single/multiple abnormalities of −7, i(17q), inv(3)/3q21, 12p−/12p11.2, 11q−/11q23, +21, or other autosomal trisomies, not including +8/+9 [eg, +21, +19]): 4 points
-
Unfavorable karyotype (all other abnormalities): 3 points
-
≥2 HMR mutations: 3 points
-
One HMR mutation: 2 points
-
Absence of CALR type 1/like mutation: 2 points
Total score, risk factor level, and median survival, and estimated 10-year survival are as follows:
-
0 points - Very low ris; median not reached (92%)
-
1–2 points - Low risk; 16.4 years (56%)
-
3–4 points; Intermediate risk 7.7 years (37%)
-
5–8 points - High risk; 4.1 years (13%)
-
≥9 points - Very high risk; 1.8 years (< 5%)
Observation alone is advised for patiens whose MIPSS70+ version 2.0 scores are in the very low and low risk categories. Allogeneic stem cell transplant is the treatment of choice for high and very high risk disease. Patients with intermediate-risk disease are best served by participation in clinical trials. [19]
GIPSS
The GIPSS (genetically inspired prognostic scoring system) is based exclusively on genetic markers: mutations and karyotype. [20] Adverse factors and scores are as follows:
-
VHR karyotype – 2 points
-
Unfavorable karyotype – 1 point
-
Absence of type 1/like CALR mutation – 1 point
-
Presence of ASXL, SRSF2, and U2AF1Q157 mutations – 1 point
GIPSS risk categories and median survivals (5-year survival rate) are as follows:
-
Low (0 points): 26.4 years (94%)
-
Intermediate-1 risk (1 point): 8.0 years (73%)
-
intermediate-2 (2 points): 4.2 years (40%)
-
High (≥3 points): 2 years (14%)
Tefferi et al favor starting determination of prognosis with the GIPSS: patients with low-risk disease can receive long-term observation, while those with high-risk disease are candidates for allogeneic stem cell transplantation. MIPSS70+ version 2.0 can be considered for confirming the most appropriate treatment approach for an individual patient. [19]
-
Primary myelofibrosis. Peripheral smear shows teardrop red blood cells (RBCs) and a leukoerythroblastic picture with nucleated RBC precursors and immature myeloid cells. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.
-
Bone marrow biopsy from a patient with primary myelofibrosis shows extensive fibrosis. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.
-
Reticulin stain on a bone marrow biopsy from a patient with primary myelofibrosis shows extensive fibrosis. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.
-
Extramedullary hematopoiesis in the spleen of a patient with primary myelofibrosis. Courtesy of Wei Wang, MD, and John Lazarchick, MD, Department of Pathology, Medical University of South Carolina.







