Practice Essentials
Factor XIII (FXIII), which was initially termed fibrin stabilizing factor, is involved in clot preservation. FXIII also participates in other physiologic processes, including wound repair and healing. FXIII deficiency, an autosomal recessive disorder, is a rare but potentially life-threatening cause of a hemorrhagic diathesis. Paradoxically, alterations in FXIII may also predispose to thrombosis.
Congenital FXIII deficiency is due principally to defects in the catalytic A subunit of FXIII, with more than 100 mutations throughout the factor XIII A gene identified. [1] Acquired FXIII deficiencies, which result from autoantibodies against FXIII subunits, are extremely rare but may produce severe bleeding diatheses. [2]
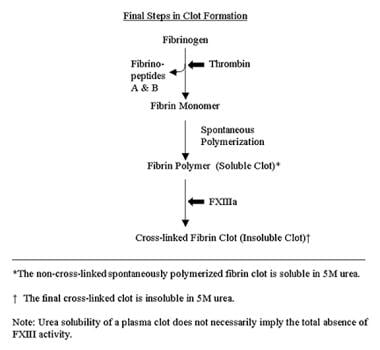
Thrombin, generated by reactions initiated by activated tissue factor VII/factor IX pathways, leads to clot formation. See the image below.
Signs and symptoms
In most cases, FXIII deficiency presents in the neonatal period. Congenital FXIII deficiency may be first evident with a wide range of bleeding manifestations. [3] Mild cases may not become apparent until trauma or surgery triggers a bleeding complication; patients with more severe deficiency may experience spontaneous bleeds, or bleeding after normal activity such as strenuous exercise. [4]
The following should trigger an evaluation for FXIII deficiency [5] :
-
Bleeding from the umbilical cord 1-19 days after birth
-
Easy bruising and soft tissue bleeding, particularly in association with trauma, as the infant starts to ambulate; bleeding following trauma may be immediate, delayed, and/or recurrent
-
CNS hemorrhage is common, recurs in approximately 30% of patients, and may be the initial manifestation in patients with severe FXIII deficiency
-
CNS bleeding may be preceded by head trauma in children, while adults may develop a CNS bleed in the absence of obvious trauma
-
Signs and symptoms typical of any CNS event may be present (eg, headaches, seizures, vomiting, focal neurologic defects); symptoms may be acute at onset or may be superimposed on residual findings of a past bleed
-
Menorrhagia and intra-abdominal bleeding during menses
-
Spontaneous miscarriages early in pregnancy
-
Bleeding into joints
-
Poor wound healing, although described, is less common
-
Autoantibodies to FXIII are an acquired cause of a bleeding diathesis; these may be triggered by isoniazid, so a detailed drug history is essential
-
Therapeutic plasma exchange can result in acquired FXIII deficiency; it may be underdiagnosed, as routine coagulation tests remain unaltered, but significant bleeding may ensue, especially in patients who have undergone recent surgery, such as a kidney transplant. [6]
Bleeding into joints
-
May be precipitated by trauma
-
Reports exist of recurrent target joint bleeds, but destructive changes in the joints are uncommon [7]
-
Spontaneous joint and extensive muscle bleeding, characteristic of patients with severe hemophilia, are uncommon in patients with severe FXIII deficiency
Physical findings
Physical findings depend on the site at which bleeding develops and include the following:
-
Bleeding from the umbilical cord after birth usually manifests with persistent oozing, which may start a few days after birth
-
Findings associated with CNS bleeding depend on the location of the bleeding; trauma may precede the event, with additional findings, or manifestations of a new CNS bleed may be superimposed on residual findings related to a prior bleed
-
Common manifestations include chronic epistaxis, bleeding gums, ecchymoses, hematomas, and periarticular bleeding; however, it is uncommon to find the large hematomas or joint bleeds characteristic in patients with severe hemophilia
-
Female patients may present with vaginal spotting or bleeding during early pregnancy, preceding a spontaneous miscarriage
-
Persistent, delayed, or recurrent bleeding may occur at sites of trauma or surgery
-
Poor wound healing may be noted
-
Acquired causes of FXIII deficiency, such as disseminated intravascular coagulation and liver disease, present in a well-recognized manner
See Presentation for more detail.
Diagnosis
The following routine tests are the first step in the evaluation of any bleeding disorder:
-
aPTT
-
PT
-
Thrombin
-
Clottable fibrinogen level
-
Platelet count
-
Bleeding time (after ascertaining that the patient was not on antiplatelet drugs for at least the preceding 5 d)
However, these tests cannot be used to screen for FXIII deficiency because the results would be within reference ranges in a patient with isolated severe FXIII deficiency. [8]
Qualitative screening test for severe FXIII deficiency
-
Assessment of clot solubility in 5M urea or 1% monochloroacetic acid
-
Lysis of thrombin and Ca2+ - induced clot within a few hours suggests severe FXIII deficiency, provided that fibrinogen levels are qualitatively and quantitatively within reference range
-
The thrombin-clottable fibrinogen test can be used to exclude hypofibrinogenemia and dysfibrinogenemia, which cause false-positive results on the 5M urea solubility test
Quantitative testing
If the 5M urea solubility test demonstrates positive results, this finding should be confirmed by quantitating FXIII activity using a monodansylcadaverine or putrescine incorporation assay.
A sensitive assay used to quantitate FXIII activity is based on monitoring the amount of ammonia (NH3) released by using glutamate dehydrogenase and nicotinamide adenine dinucleotide phosphate during the transamidation reaction (cross-linking) by FXIII. Another sensitive colorimetric assay is based on incorporation of 5-(biotinamido) pentylamine into fibrin/fibrinogen. [9]
In addition, a2PI and plasminogen activator inhibitor–1 assays should be performed to exclude abnormalities in the fibrinolytic pathway, which accelerate clot lysis. Sodium dodecylsulfate polyacrylamide gel electrophoresis under reducing conditions has been used to assess the presence of cross-linked g or a chains of fibrin, which is a reflection of FXIII activity. The studies must be performed by laboratory personnel with special expertise.
Testing for inhibitors
-
Repeat of the urea solubility test with mixtures containing varying proportions of patient and normal plasma to differentiate between a deficiency or an inhibitor as the cause of a positive result; serum may be substituted for plasma in the test
-
Semiquantitation of the susceptibility of the fibrin clot to fibrinolysis can be obtained by adding iodine-125-labeled fibrinogen, tissue plasminogen activator, thrombin, and Ca2 + to the patient's plasma, with measurement of the time to 50% clot lysis
Prenatal diagnosis
-
Chorionic villous sampling at approximately 10-12 weeks of gestation or amniocentesis at 16-20 weeks of gestation can be performed to obtain fetal cells for DNA analysis or for linkage studies.
-
If DNA analysis cannot be performed, fetal blood obtained by fetoscopy at approximately 20 weeks of gestation can be used.
-
Perform these procedures only after intense genetic and obstetric counseling of the parents.
See Workup for more detail.
Management
FXIII replacement is used to treat bleeding, to prevent perioperative bleeding during elective surgical procedures or, prophylactically, to prevent recurrent bleeding, as in CNS or joint hemorrhages. Serial monitoring of achieved FXIII levels is essential to document the adequacy of any therapy.
FXIII concentrates for replacement are as follows:
-
Plasma-derived virus-inactivated human FXIII concentrate (Corifact, in the United States; Fibrogammin P in Europe, South America, South Africa, and Japan); a second FXIII concentrate (Bio Products Laboratory, Elstree, Hertfordshire, UK) is available on a per-patient request
-
Recombinant FXIII A-subunit, recombinant (Tretten) [10]
Minor bleeding, as from cuts and abrasions, may respond to conservative measures, such as pressure, ice, and use of antifibrinolytic drugs. Avoidance of trauma and nonsteroidal anti-inflammatory drugs (NSAIDs) is helpful in reducing bleeding events.
Treatment of patients with inhibitors
-
FXIII dose depends on the characteristics of the inhibitor
-
Also treat the underlying disorder and, when appropriate, use immunosuppressive agents, including B-cell–directed monoclonal antibodies.
-
Note that spontaneous disappearance of acquired inhibitors is part of their natural history, and the use of milder, less toxic immunomodulators, such as steroids, may suffice
-
Simple immediate ancillary measures of ice, pressure, elastic bandages, immobilization of the affected joint, and avoidance of NSAIDs may suffice in some cases
See Treatment and Medication for more detail.
Background
The presence of a bleeding diathesis in families with an X-linked pattern of inheritance of the disorder has been recognized for hundreds of years. The recognition of factor deficiencies as the cause of hemophilias spurred investigations into the causes of other bleeding disorders and led to progress in understanding normal hemostasis. Knowledge of the fact that blood clots formed in the presence of calcium are stronger, insoluble in alkali, and resistant to proteolytic degradation led to the concept of insoluble clots in the early 20th century.
In 1948, Laki and Lorand recognized that a serum factor, termed fibrin stabilizing factor, was responsible for the characteristics of insoluble fibrin clots. [11] In 1960, Duckert et al described the first case of an "undescribed congenital haemorrhagic diathesis probably due to fibrin stabilizing factor deficiency," which was a description of the consequences of severe factor XIII (FXIII) deficiency. [12, 13] In 1963, the International Committee on Blood Clotting Factors recognized fibrin stabilizing factor as a clotting factor and named it factor XIII. [14]
The importance of FXIII in the process of coagulation is underscored by symptoms borne by patients who are homozygously deficient in FXIII or who have an antibody that disrupts FXIII function. Paradoxically, alterations in FXIII may predispose patients to thrombosis. Based on all available data, FXIII is clearly involved in the clot preservation side of the delicate balance between clot formation and stability and clot degradation. FXIII participates in other physiologic processes, including wound repair and healing.
Gene polymorphisms are being evaluated for their influence on susceptibility to venous and arterial thromboembolism. [15] Variants of coagulation factors, including FXIII Val34Leu, have been implicated in influencing susceptibility to thromboembolic diseases. [16]
There is a question as to whether FXIII Val34Leu polymorphism is protective against idiopathic venous thromboembolism.The substitution of leucine for valine at amino acid position 34 of the FXIII gene, commonly referred to as FXIII Val34Leu polymorphism, has been reported to confer protection against venous thromboembolism. However, the results of a study in a White Canadian population did not support an independent association of the FXIII Val34Leu polymorphism with idiopathic venous thromboembolism. [17]
An association may exist between the FXIII Leu allele and a modest protective effect against acute myocardial infarction (MI). [18, 19] However, young women with the Leu34/Leu34 genotype have been found to have a nearly fourfold higher risk of ischemic stroke compared with those with the Val34/Val34 genotype, [19] and FXIII Val34/Leu polymorphism has been associated with increased coronary disease (CAD) risk, although not with CAD without MI. [20]
FXIII measurement has a variety of uses, potential and confirmed. Plasma levels of FXIII were found to be decreased in children with Henoch-Schönlein purpura having severe abdominal symptoms. Thus, it has been suggested that measurement of FXIII level may be of value to detect the vasculitic process of Henoch-Schönlein purpura before the rash occurs or long after it has disappeared in patients with isolated abdominal or scrotal problems. [21, 22] Immunohistochemistry studies have shown that FXIIIa-positive dermal dendritic cells were increased in a variety of skin tumors, including dermatofibromas. [23]
Severe FXIII deficiency, a rare autosomal recessive coagulation disorder, is associated with a relatively common prevalence of F13B gene defects, at least within the German population. The regions in and around the cysteine disulphide bonds in the FXIII-B protein are the sites of frequent mutations. [24] It is relatively common in Iran, especially in Khash; most patients there have a unique mutation in the F13A gene (Trp187Arg), producing severe FXIII deficiency. [25]
FXIIIs aids immobilization and killing of bacteria as well as phagocytosis by macrophages, likely functioning as part of the innate immune system. [26]
Use of relatively new specific FXIII assays are pivotal to avoid missing the diagnosis of FXIII deficiency, a rare but potentially life-threatening disorder. [27]
Pathophysiology
Structure, production, and half-life of FXIII
Plasma FXIII is a heterotetramer consisting of 2 identical proenzyme subunits (A2) and 2 identical carrier protein subunits (B2). Subunit A contains the catalytic site, the activation peptide, a calcium-binding site, and free sulfhydryl (SH) groups. Subunit B, a glycoprotein, acts as a carrier protein that stabilizes subunit A, binds the zymogen (subunit A) to fibrinogen, and acts as a brake on FXIII activation. [28, 29] Subunit B circulates in plasma as part of the tetramer A2 B2 and as a free B2 dimer; all of plasma subunit A is complexed with subunit B. The concentration of subunit A in plasma is 15 mg/mL, while that of subunit B is 21 mg/mL. Much of FXIII circulates in blood in association with fibrinogen. [30, 31]
Platelet FXIII (an A2 homodimer) constitutes approximately 50% of total FXIII activity in blood. Plasma FXIII has a long half-life of approximately 9-14 days. A similarity exists between a portion of the carboxy terminal (C terminal) domain of FXIII and the receptor-binding region of a2 -macroglobulin. The complex of a2 -macroglobulin and its substrate protease is removed from the circulation by binding to its receptor in the liver and other tissues; therefore, as has been suggested, FXIII also may be removed from the circulation by a similar mechanism. [32, 33] Some features of the A and B chains of FXIII are listed below. Monoclonal antibodies and naturally occurring inhibitors are used to elucidate structure-activity relationships.
Bone marrow cells, megakaryocytes, and monocytes/macrophages synthesize FXIII, with a possible role for hepatocytes in the synthesis of subunit A. Subunit B is synthesized by the liver. Tissue transglutaminase, the intracellular form of FXIII, consists of the A2 subunit (an A2 homodimer) and is present in a variety of cells including platelets, megakaryocytes, monocytes/macrophages, and in the liver, placenta, uterus, prostate, and dermal dendrocytes. [34] Red cells contain a transglutaminase that is activated by Ca2+ but is different from plasma transglutaminase in its cross-linking activity and can cross-link fibrinogen as well as fibrin. Trapped erythrocytes release FXIII when red cells lyse, providing additional cross-links to the aging thrombus. [28]
Table. Some Features of the A and B Chains of Factor XIII (Open Table in a new window)
Properties |
A Chain |
B Chain |
Plasma FXIII |
Has 2 A chains |
Has 2 B chains |
Plasma level |
Approximately 15 mg/mL |
Approximately 21 mg/mL |
Chains are free in plasma |
No. All bound to B chain and present as an A2 B2 tetramer |
Yes. Excess B chain present in plasma as a B2 dimer |
Chain contains the catalytic site |
Yes |
No |
Chain is the carrier protein |
No |
Yes |
Chain acts as a brake on FXIII activation |
No |
Yes |
Cellular FXIII |
Has 2 A chains (A2 dimer) |
Has no B chains |
Mutations can lead to decreased FXIII activity |
Yes |
Yes |
Comparative biology shows that transglutaminases are distributed widely in nature and may represent the prototype for the evolution of clotting enzymes. [35] Partial homology of plasma FXIII exists with several proteins including tissue and keratinocyte transglutaminases, erythrocyte transglutaminase, and the hemocyte transglutaminase of the horseshoe crab and other zymogens of the same family.
A recent example is from the crystal structure of transglutaminase of the Red Sea bream, which shows that its active site and overall structure resemble that of human FXIII. [36] These homologies attest to conservation of the enzyme during evolution. Since the gene structures are similar, it is believed that they evolved from a common ancestor. Subunit B contains 10 repeating "sushi" units linked by disulfide bonds; the function of the sushi unit is unknown. Sushi structures are present in at least 26 proteins, including proteins in the horseshoe crab and in the vaccinia virus.
Activation
Thrombin, generated by reactions initiated by activated tissue factor VII/factor IX pathways (as illustrated in the first diagram below), leads to clot formation. Thrombin releases fibrinopeptide A from the a chain of fibrinogen, then fibrinopeptide B from the b chain of fibrinogen. Fibrin monomers (formed following the release of fibrinopeptides) polymerize spontaneously; this is followed by development of a complex branching clot as a result of the actions of activated FXIII (FXIIIa). [37] The sequence of these final steps is found in the second chart below.
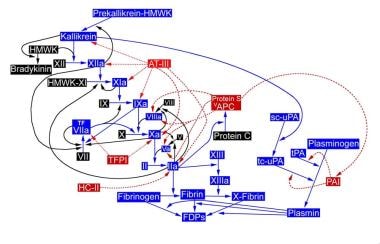 Coagulation reactions leading to thrombin generation and activation of factor XIII.
Coagulation reactions leading to thrombin generation and activation of factor XIII.
Thrombin starts the process of FXIII activation by cleaving an activation peptide from subunit A. The subsequent Ca2+ -dependent dissociation of subunit B allows FXIII activation to proceed. Calcium is important for activation of the zymogen (both FXIII and tissue transglutaminase require Ca2+), conformational changes, and opening of the catalytic site of FXIII to its substrate. Calcium also provides physical stability as determined by x-ray crystallography, computer modeling, and other studies; all of the changes allow the active subunit A to perform its functions optimally. [28, 38, 39]
When activated by thrombin, tissue FXIII functions in the same manner as plasma FXIIIa. Platelet FXIII undergoes nonproteolytic activation following the platelet activation-induced rise in cytosolic Ca2+. Activation of the red cell enzyme occurs upon exposure to Ca2+, and red cells that are present in the fibrin clot lyse and release their FXIII as the clot ages. Several controls in the complex activation process focus the actions of FXIIIa on fibrin rather than on fibrinogen. Cross-linking of polymerized soluble fibrin by FXIIIa is the final step in hemostasis, as illustrated in the image below. For extensive details of this activation process, the reader is referred to two recent reviews by Lorand. [28, 35]
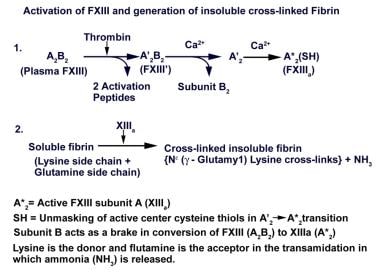 Activation of factor XIII and generation of insoluble cross-linked fibrin. Adapted from Lorand L. Ann N Y Acad Sci. 2001;936:291-311.
Activation of factor XIII and generation of insoluble cross-linked fibrin. Adapted from Lorand L. Ann N Y Acad Sci. 2001;936:291-311.
Role of FXIII in cross-linking and resistance to lysis
FXIIIa cross-links the lysine of one g chain in the fibrin polymer with the glutamine of another g chain by transamidation, releasing ammonia in the process. Additional cross-links occur between a-a chains, a-g chains, a chains-a2 -plasmin inhibitor (a2 PI), and a chains-fibronectin. As a result of the extensive cross-linking actions of FXIIIa, the clot structure of fibrin polymers increases in complexity from dimers to trimers to tetramers.
The g chains of fibrinogen and fibrin normally bind to the platelet membrane glycoprotein IIb/IIIa complex. The same g chains are subject to cross-linking by FXIIIa; therefore, cross-linking also occurs between fibrin and the platelet membrane. Both plasma FXIIIa and platelet FXIIIa cross-link fibrin polymers, but under physiologic conditions, platelet FXIII is believed to play a minor role. [40] Red cell FXIII is responsible for hybrid cross-linking of a-g chains, in contrast to the actions of plasma FXIII.
Dysfibrinogenemias and dyshypofibrinogenemias result in alterations in fibrin (substrate for FXIIIa), which can interfere with the ability of FXIII to cross-link fibrin. A reduction in available fibrin resulting from afibrinogenemia can have the same effect. Conversely, increased fibrinogen levels have been identified as a risk factor for thrombosis.
Mechanisms of this risk were elucidated by a fibrinolysis assay containing purified components. The assay showed that lysis of fibrin decreased as fibrinogen levels increased, and the presence of a minor common variant of fibrin (g') is associated with accelerated cross-linking, which made the clot resistant to proteolysis by both plasmin and trypsin. Increased clot stability also was believed to result from increased concentration of FXIII in the clot. Non-cross-linked fibrin potentiates activation of FXIII by thrombin; thus, the substrate potentiates its enzyme, further contributing to clot stability. [31, 41, 42]
Cross-linking of a2 PI to a chains of fibrin by FXIIIa brings the principal inhibitor of plasmin to the site of the clot, ensuring resistance of the clot to proteolysis. Inhibition of a2 PI in in vitro systems leads to enhanced clot lysis. In humans, deficiency of a2 PI results in a bleeding disorder because of vulnerability of the fibrin clot to prompt degradation by plasmin. The formation of highly cross-linked a-fibrin polymers in the presence of high concentrations of FXIIIa produces clots that are highly resistant to fibrinolysis. [43]
Fibronectin, an adhesive protein, is a large component (approximately 4%) of the proteins in a fibrin clot, is present in plasma and cells, and is subject to cross-linking by both plasma and cellular FXIII. Cross-linking of fibronectin to fibronectin and fibronectin to fibrin is accomplished by FXIIIa, with fibronectin contributing to increased fiber size, density, and strength of the clot. FXIIIa also cross-links actin to fibrin and actin to myosin. Cross-linking of intracellular structural proteins is involved in clot retraction and cell migration. This complex gel network created by the actions of FXIII plays an important role in wound healing, cell adhesion, and cell migration. All of these cross-linking reactions impart increased mechanical strength to the clot, contributing to clot retraction and resistance of the clot to degradation by plasmin and providing an explanation for the known plasmin resistance of older clots.
Many other proteins function as substrates for FXIIIa, including von Willebrand factor (vWF), factor V (FV), thrombospondin, gelsolin, vitronectin, vinculin, lipoprotein (a), and collagen (FXIIIa cross-links collagen with fibronectin and vWF, attaches the clot to the vessel wall, impacts tissue repair, increases resistance of collagen to proteolysis, and modulates synthesis of collagen by fibroblasts). Thus, FXIII plays a role in a wide array of cross-linking reactions involving plasma proteins at the intracellular level, impacting many different functions.
Note that because of the lack of cross-linking in individuals with FXIII deficiency, their D-dimer level will remain low, even if they experience thrombosis, although levels of other fibrin degradation products will be increased. Consequently, the D-dimer assay is not reliable for screening in these patients.
Factors affecting level and activity of FXIII
When quantitative amine incorporation assays became available, healthy people were found to have an 8-fold spread in FXIIIa activity. [44] In recent studies of FXIII antigen and activity in humans, no correlation was found between these two parameters. During the search for an explanation, 23 unique FXIIIa genotypes were found. The Leu34 and Leu564 variants gave rise to increased specific activity; the Phe204 variant lowered specific activity. Other mutations gave rise to low, high, or median FXIII-specific activity, and some variants had no effect. [45, 46]
In a study of the variability of FXIII levels in racial groups, FXIII activity was found to be higher in Asian Indians (male and female) than in their Chinese counterparts, accounting for approximately one fourth of the variability. Common genetic polymorphisms in the A and B chains appeared to contribute to the differences. [47] An influence exerted by acquired factors was evident in the higher FXIII levels found in women who smoked 20 or more cigarettes per day during a normal pregnancy than was found in nonsmokers, with a lesser drop in the second half of pregnancy. [48]
Role of FXIII in pregnancy
In the latter half of pregnancy, some drop in FXIII levels is normal, but severe (homozygous) FXIII deficiency is a cause of recurrent miscarriages. In a study of gestational tissues, FXIII was found in the decidual layer of the placenta, while FXIII secretion was evident in cultures of round-shaped endometrial cells. A study of early (7-8 wk) gestational tissues obtained from women without FXIII deficiency and from a woman who was homozygous for FXIII deficiency showed poorly formed cytotrophoblastic shells and Nitabuch layers, along with absence of FXIIIa in tissues obtained from the woman with FXIII deficiency. Low plasma levels of FXIII appear to correlate with low placental levels of FXIII with poor trophoblastic development, which may be the cause of spontaneous miscarriages.
It has been suggested that preventing miscarriage in patients who are severely deficient requires FXIII supplementation beginning at approximately 5 weeks of gestation because FXIII, fibrinogen, and fibronectin are necessary to anchor cytotrophoblasts invading the endometrium. [49, 50] Reduced FXIII activity resulting from the Tyr204Phe mutation has been associated with repeated miscarriages. [51]
Role of FXIII in wound healing
Physiologically, hyperpermeability induced by severe metabolic inhibition of porcine aortic endothelial cells is prevented by FXIIIa, which is similar to the maintenance of endothelial barrier function by FXIIIa despite depletion of energy or during reperfusion of ischemic rat hearts. [52] In a different system, FXIII induced epithelial wound healing by increasing cell growth by approximately 2.5 fold, leading to replacement of damaged cells. [53] Smooth muscle cell migration, an integral part of the healing process, is facilitated by FXIII. Migration of smooth muscle cells in cross-linked fibrin gels was twice the migration seen in non–cross-linked gels, demonstrating the importance of the 3-dimensional clot structure created by cross-linking in smooth muscle cell migration. [54] In humans, Fibrogammin was shown to contribute to the healing of venous leg ulcers by reducing endothelial permeability. [55]
Effects of other agents on FXIII
Nitric oxide (NO), an important diffusible molecular messenger, is increasingly recognized as having an impact on coagulation proteins. Activity of plasma transglutaminase is inhibited by NO via nitrosylation of critical thiol groups (reactive cysteine residue), resulting in inhibition of both g-chain cross-linking and insoluble clot formation. NO donors and carriers inhibit FXIII activity in a dose-dependent manner, in a purified system and in plasma. Tissue transglutaminases are involved in apoptosis, and inhibition of their activity by NO prevents apoptosis. [56, 57]
Venoms and toxins can affect clot stability. Excessive bleeding resulting from envenomation can affect the functions of FXIII in different ways. Acuthrombin A, one of two proteases in the venom of Agkistrodon acutus (five-pace snake), activates FXIII. [58] Ancrod, obtained from the venom of Agkistrodon species, causes defibrination, thereby removing the substrate for FXIII.
A severe systemic bleeding disorder may develop several hours after initial contact with 2 types of caterpillars in the Saturniidae family (from Brazil and Venezuela). Intracranial and intracerebral bleeding and renal failure may follow. In this case, FXIII reduction results from generalized disseminated intravascular coagulation (DIC) induced by several activities directed against the hemostatic mechanism, including a FXIII proteolytic-urokinase–like activity. [59] Tridegin, a peptide inhibitor of FXIII present in the saliva of an Amazon leech (Haementeria ghilianii) accelerates fibrinolysis by inhibiting FXIIIa; tridegin is under investigation as a potential new antithrombotic agent. Destabilase, an enzyme present in the leech, hydrolyzes g-g fibrin cross-links and breaks down blood clots. [60]
Simvastatin is a commonly used cholesterol-lowering agent. A non–antibody-mediated drug-induced reduction in FXIII activity as part of a broader reduction in hemostatic activation has been suggested to be the reason for the proven antithrombotic efficacy of simvastatin in clinical trials. [61] Blood samples were obtained sequentially every 30 seconds from a bleeding time cut in patients with coronary artery disease, before and 3 months after simvastatin treatment. Samples were analyzed for the time-course drop in fibrinogen levels and activation of factors II, V, and XIII by quantitative Western blot analyses. Simvastatin, independent of its effects on cholesterol, significantly reduced the rate of blood clotting, as evidenced by reduced formation of several activation products including FXIIIa.
Several selective synthetic inhibitors have been shown to prevent the ability of FXIIIa to stabilize a clot, thereby reducing clot strength (clot stiffness, viscoelastic modulus) to approximately 20% of normal (values similar to those seen in patients with severe FXIII deficiency). Rapid lysis of these clots occurred following in vitro exposure to thrombolytic agents. [35] Imidazolium derivatives, a new class of compounds, specifically inhibit both FXIII-induced formation of a-chain polymers and the incorporation of a2 PI into the a chain of fibrin, resulting in accelerated clot lysis. [62, 63]
Specific monoclonal antibodies to FXIII have provided similar benefits by reducing the viscoelastic properties and by enhancing clot lysis. They also have been used to modify disease states. The beneficial effect of the absence of cross-linked fibrin on pathophysiologic processes was proven in an animal model of widespread thrombosis. FXIIIa deficiency induced in rabbits by pretreatment with a specific monoclonal antibody before induction of a generalized Schwartzman reaction protected them from the deleterious effects of widespread microvascular thrombosis. The protection resulted from the ability of the fibrinolytic system to effectively degrade non–cross-linked thrombi. [64] These data add support to the author's speculation many years ago of the potential use of drugs that inhibit cross-linking as a method of prophylaxis in venous thromboembolic disease.
The biochemical basis and potential for using modifiers of fibrin stabilization in improved thrombolytic therapies are discussed in a recent review by Lorand. [35] Similar ideas have been proposed by others, expanding on the importance of fibrin structure in thrombus formation and dissolution. [65] Prospective clinical trials must prove any thromboprophylactic efficacy of altering fibrin structure using specific drugs.
Other functions of FXIII
Plasma and tissue transglutaminases have been reported to promote cell adhesion through specific integrins for 2 different tumor cell types, MOLT-3 human lymphocyte–like leukemia and melanoma cells and SW480 colon cancer cells transfected with a ligand. [66] In contrast, FXIII did not stimulate growth of cultured human tumor cells. [67] An intriguing observation is the potential use of subunit A of FXIII and FXIII activity as a tumor marker in malignant brain tumors; its presence may distinguish benign from malignant brain tumors. [68] Recently, it was shown for the first time that intranuclear accumulation and cross-linking activity of FXIIIa occurred in maturing monocytes, supporting the hypothesis that FXIIIa may be involved in cell proliferation/differentiation, chromatin structure remodeling, and even cell death. [69] Further data are needed to unravel the role of FXIII in malignancies.
An unexpected role has been postulated for FXIII in degenerative brain disorders. In Alzheimer disease and spongiform encephalopathies, the brain contains fibrils that develop from native proteins containing a discordant a helix. Human FXIII was found to form fibrils in buffered saline, suggesting that FXIII, in addition to several other proteins, can be a source of this abnormal fibrillar protein. [70]
Possible interactions between deficiencies of FXIII and thrombin-activatable fibrinolytic inhibitor
Thrombin-activatable fibrinolytic inhibitor (TAFI), a single-chain carboxypeptidase B–like zymogen, is activated by thrombin to become activated TAFI (TAFIa). [71] The importance of TAFIa in fibrinolysis is emphasized by the fact that the conversion of only 1% of the zymogen to TAFIa is sufficient to suppress fibrinolysis by approximately 60%.
TAFIa suppresses fibrinolysis by removing C-terminal lysine and arginine residues exposed in the partially degraded fibrin clot produced by plasmin. Removal of C-terminal lysine residues from fibrin reduces the rate of plasminogen activation by a number of mechanisms, attenuating fibrinolysis. This effect is counterbalanced in normal plasma by activation of protein C, which has profibrinolytic properties because of its ability to suppress thrombin generation via its major effect of degrading activated factor V (FVa), and to a lesser extent, activated factor VIII (FVIIIa). [71, 72, 73]
As illustrated in the chart below, a delicate balance usually exists between thrombus formation and thrombus resolution; thrombin secures survival of the thrombus created by its action on fibrinogen by activating TAFI, thereby inhibiting fibrinolysis. Cross-linking of fibrin induced by FXIIIa (activated by thrombin) renders the clot insoluble. FXIII deficiency results in absence of cross-linked fibrin, leading to premature lysis of the clot by the fibrinolytic system; adverse consequences result, including bleeding. Theoretically, a deficiency of TAFI leading to decreased suppression of fibrinolysis (enhanced clot lysis) can potentiate bleeding resulting from FXIII deficiency (also associated with enhanced clot lysis). Note the image below.
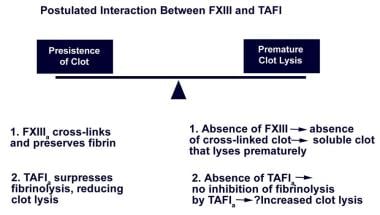 Postulated interaction between factor XIII and thrombin-activatable fibrinolytic inhibitor.
Postulated interaction between factor XIII and thrombin-activatable fibrinolytic inhibitor.
Cell surface–directed hemostasis
The concept of coagulation as a waterfall or cascade effect has been acknowledged for a long time, with platelets and other cell surfaces providing the anionic phospholipids needed for complex formation, so that reactions can proceed efficiently. One review proposed that coagulation is essentially a cell surface–based event. [74] Platelet FXIII is positioned appropriately to influence the process. (See the diagram below.)
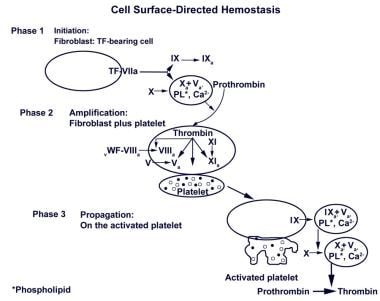 Cell surfaced–directed hemostasis. Initially, a small amount of thrombin is generated on the surface of the tissue factor–bearing (TF-bearing) cell. Following amplification, the second burst generates a larger amount of thrombin, leading to fibrin (clot) formation (from article: Factor XIII). Adapted from Hoffman and Monroe. Thromb Haemost. 2001;85(6):958-65.
Cell surfaced–directed hemostasis. Initially, a small amount of thrombin is generated on the surface of the tissue factor–bearing (TF-bearing) cell. Following amplification, the second burst generates a larger amount of thrombin, leading to fibrin (clot) formation (from article: Factor XIII). Adapted from Hoffman and Monroe. Thromb Haemost. 2001;85(6):958-65.
Conclusion
Much work is needed, even in the clinical arena, to clarify the relationship between the exact levels of FXIII and hemorrhagic or thrombotic phenotypes. Establishing an international registry of patients deficient in FXIII would be of value in improving understanding of the protean manifestations of this uncommon disorder.
Etiology
To date, most identified mutations in patients with severe FXIII deficiency and a bleeding disorder involve subunit A, with very few mutations reported involving subunit B. The gene for subunit A is located on chromosome 6, bands p24-25. The gene is 160 kilobases in length and has 15 exons and 14 introns with specific structural and functional domains. Catalytic activity is encoded in the second exon, and the active cysteine is encoded by the seventh exon. The 2 Ca2+-binding sites and a thrombin-inactivation site have been identified at other locations. The gene for subunit B is located on chromosome 1, bands q31-32.1, is 28 kilobases in length, and has 12 exons and 11 introns. [75, 76] (See the image below.)
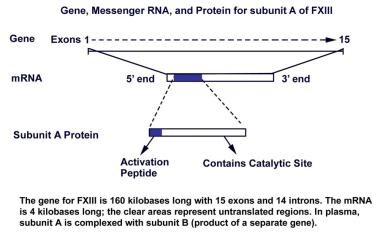 Gene, messenger RNA, and protein for subunit A of factor XIII. Adapted from Reitsma PH. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Lippincott Williams & Wilkins; 2001:59-87 and from Roberts HR, Monroe DM III, Hoffman M. In: Williams Hematology. McGraw-Hill Professional; 2001:1409-34.
Gene, messenger RNA, and protein for subunit A of factor XIII. Adapted from Reitsma PH. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Lippincott Williams & Wilkins; 2001:59-87 and from Roberts HR, Monroe DM III, Hoffman M. In: Williams Hematology. McGraw-Hill Professional; 2001:1409-34.
Detailed characteristics of complementary RNA (cRNA) and messenger RNA (mRNA) of the placental subunit A are known. The presence of an acetylated amino terminal end and the absence of glycosylation and disulfide bonds apparently are features typical of secreted cytoplasmic proteins. The presence of these characteristics makes it conducive for subunit A expressed in yeast systems to make a recombinant product.
Substitutions in the core domain of the enzyme, affecting highly conserved residues, result in a serious defect in structure and function. Missense mutations in the A chain are a common cause, accounting for approximately 50% of cases of severe FXIII deficiency. The defects result in an absence of subunit A protein but also are accompanied by a reduction in subunit B carrier protein (type II defect).
Nonsense mutations are an equally common cause of A chain defects, resulting in a frameshift-type, splice-type, or termination-type mutation. The few defects that have been reported in the B chain lead to a deficiency of the carrier protein (subunit B), which then leads to instability and reduction of plasma subunit A levels despite the presence of functional intracellular subunit A (type I defect). [77] Therefore, patients who are homozygous for subunit B mutations have a bleeding disorder. Most recently, impaired intracellular transport from the endoplasmic reticulum to the Golgi apparatus, with failure of secretion of the truncated FXIII subunit B produced by a single-base deletion, was reported to be the cause of severe FXIII deficiency in 3 unrelated patients. [78]
Many kinds of mutations have been (and continue to be) identified, with some mutations unique to certain families. The finding of compound heterozygotes has eliminated the mandatory search for consanguinity in all parents of patients with severe FXIII deficiency. [79, 80, 51]
An unusual mutation has been described in 2 Finnish sisters with a very mild bleeding disorder. One sister had 2 successful pregnancies without regular replacement therapy. The sisters had no detectable subunit A activity (< 1%) using plasma screening tests; however, using the 3H-putrescine incorporation assay, subunit A showed 0.35% of normal activity, with partial g-g dimerization of fibrin in clotted plasma. A full-length subunit A was detected in the patients' platelets using Western blot analysis.
The sisters had an Arg661→stop mutation on one allele and a T→C transition on the other allele. These data showed that a mutation in the splice donor site of intron C can result in different variant mRNA transcripts and that small amounts of correctly processed mRNA can produce a type of FXIII that can produce, at least, dimerization of fibrin, thus minimizing the clinical consequences. [81]
Various reported mutations are spread throughout the gene coding for FXIII without specific hot spots. In many patients, low steady-state mRNA levels have been found, which result in inefficient production of the abnormal protein. [62]
Data in the literature conflict regarding the impact of the common FXIII subunit A Val34Leu mutation (associated with higher plasma transglutaminase activity) on thrombotic disease. Note the following:
-
The Val34Leu mutation continues to be studied in different populations because current data provide conflicting evidence about its causal role in coronary artery disease. The Val34Leu mutation appears to protect Whites but not Asian Indians from myocardial infarction. However, in the Asian Indian population in Britain, a strong link was found between FXIII subunit B levels and risk factors for cardiovascular disease and, possibly, insulin resistance. [82]
-
A meta-analysis concluded that carriage of the Leu allele of the FXIII gene might slightly increase the risk of intracerebral hemorrhage in Whites. [83] However, a case-control study found that carriage of the FXIII-A Val34Leu polymorphism is associated with a significantly smaller clot burden in patients with ischemic stroke. [84]
A possible cooperative interaction between the Val34Leu mutation and other known thrombophilic mutations also has been explored. Note the following:
-
In a study of patients from southern France, a higher than usual odds ratio was found for the association between carriers of an angiotensin receptor mutation and coronary artery disease, but no association was found between the disease and any of the FXIII polymorphisms that were studied. [85]
-
A study of the contribution of the Val34Leu mutation to thrombotic risk in a large number of carriers of factor V Leiden (who were relatives of thrombotic probands with factor V Leiden) found a very modest contribution of the Val34Leu mutation to venous thrombotic disease. [86] This study contrasts with a report of a protective role of the mutation in venous thrombosis. [87]
-
A review by Kohler discusses the possible role of FXIII in vascular diseases. [88] The FXIII Val34Leu mutation does not appear to influence the induction or modification of the course of inflammatory bowel disease.
Genetic polymorphisms affecting both the A and B subunits have been reported, but because they do not involve conserved amino acids or are not important for protein structure, they do not result in FXIII deficiency and bleeding. Based on an analysis of polymorphisms in the gene for FXIII subunit A and their products in a northern Portuguese population, it has been stated that the evolutionary order of appearance of the main protein alleles for FXIII is 1B-->2B-->1A-->2A and that intragenic combinations are likely to have played a role in the molecular diversity in the main FXIII subunit A alleles. [89]
Genetic polymorphisms and, particularly, intragenic polymorphisms are useful in genetic counseling of families with unknown mutations. For example, 80% of whites are heterozygous for a tetrameric repeat in intron 1 of subunit A, which can help differentiate defects in subunit A from defects in subunit B. [75, 62, 76, 9] Some polymorphisms are universal, while others appear to be restricted to particular ethnic groups. The latter situation will change as ethnic intermarriages increase in this global society. Families with severe FXIII deficiency associated with a serious disabling bleeding disorder have access to all of the genetic tools available to patients with hemophilia A and B. Next-generation sequencing technology allows a rapid and simultaneous analysis of all regions of all the genes involved in the disorder. [90]
Disorders of fibrin stabilization can affect the activity of FXIII or its substrates fibrin and fibrinogen. A proposed classification of disorders leading to a positive urea solubility test result is presented below.
Abnormalities of FXIII (enzyme) are as follows:
-
Genetic mutation - (1) Subunit A, (2) subunit B, (3) subunit A and B
-
Acquired - (1) Decreased production (ie, liver disease), (2) increased loss due to excessive activation (ie, DIC, exposure to snake venoms and caterpillar toxins), (3) secondary to inhibitors (ie, alloantibodies and autoantibodies)
Abnormalities of the substrate for FXIII (fibrin/fibrinogen) are as follows:
-
Genetic mutation - (1) Afibrinogenemia, (2) dyshypofibrinogenemia
-
Acquired - (1) Decreased production (ie, acute massive hepatic necrosis or severe chronic liver disease), (2) increased loss resulting from defibrination syndromes (ie, DIC, exposure to snake venoms and caterpillar toxins, and systemic hyperfibrinolysis [drug induced or disease induced])
Epidemiology
Overall estimated frequency of the autosomal recessive disorder involving a severe deficiency of subunit A is approximately 1 case per 2 million population. [91] FXIII deficiency has been reported in many ethnic groups around the world, including persons from Canada, Europe, India, Israel, Japan, Kuala Lumpur, Pakistan, Papua New Guinea, South America, Thailand, Turkey, and the United States.
Consanguinity is not required but does increase the risk for congenital FXIII deficiency. [92] In a small series of 20 children from 16 families in India, the rate of consanguinity was 75%. [93] A high rate of consanguinity was also documented in affected persons in Iran. [94] In Khash, Iran the prevalence is one homozygote per approximately 500 population, which is higher than its worldwide prevalence, with 3.5% heterozygotes. [25]
A prospective multicenter cohort project of inherited bleeding disorders in France identified 10,047 patients, with only 5.0% having a clotting factor deficiency of the uncommon varieties, specifically Factor I, II, V, combined V and VIII, VII, X, XI, and XIII. [95]
Diagnosis of disorders of FXIII inhibitors, which may have been missed in the past, is increasing as more laboratory support becomes available around the world. Increasing use of isoniazid (INH) to combat a worldwide rise in incidence of tuberculosis could contribute to an increased incidence of FXIII inhibitors in patients.
Variability in the distribution of mutations is exemplified by existing data. For example, significant ethnic heterogeneity was found in a Brazilian population in which the Val34Leu mutation was present in 51.2% of South American Indians, 44% of Whites, and 28.9% of Blacks, but in only 2.5% of Japanese Asians. [96]
Race-, sex-, and age-related demographics
No racial predilection exists for FXIII deficiency. FXIII deficiency has been reported widely. The restriction of certain polymorphisms to specific populations should be expected.
Since it is an autosomal disorder, homozygous FXIII deficiency occurs in either sex. Acquired inhibitors to FXIII can present in either males or females.
Physiologically, reduced levels of FXIII are found in healthy newborns, with a gradual rise in levels into the reference range. Premature infants have lower values than full-term neonates. FXIII levels drop in the latter half of a normal pregnancy.
Neonates with severe FXIII deficiency may present with bleeding from the umbilical cord. Easy bruising and delayed and recurrent bleeding after trauma begin in childhood. Oral bleeding can begin with teething and cuts or abrasions to the lips, tongue, and frenulum. Bleeding remains a problem throughout life and requires replacement therapy. FXIII deficiency acquired as a result of autoantibodies has been reported in the older population, as has acquired hemophilia A. Both drug-induced autoantibodies and alloantibodies have been reported in severely deficient patients who have been receiving replacement therapy.
Prognosis
Prognosis depends on the types of complications that develop, on the type of replacement product the patient has received, and on the viral infections that the patient has accumulated over the years. Newly diagnosed patients should, whenever possible, receive purer products to ensure maximum safety. The presence of inhibitors in patients poses a serious therapeutic challenge, and, currently, surgery should be considered only as a lifesaving measure.
Umbilical bleeding starting in the first few days after birth, recurrent intracranial bleeding, and recurrent early miscarriages are hallmarks of FXIII deficiency. Pregnant women with FXIII deficiency have a significant risk of miscarriage, placental abruption, and postpartum hemorrhage without prophylaxis. [97]
Approximately 30% of central nervous system (CNS) bleeding is recurrent, and approximately 50% of CNS bleeding may be fatal, but the severity of bleeding varies from family to family. It may be of special concern for infants, particularly after trauma. [98] Posttraumatic bleeding may be immediate, delayed, or recurrent. Traumatic joint bleeding may develop. Poor wound healing has been described, although this is not a universal finding.
Cryoprecipitate and fresh frozen plasma (FFP) provide a source of FXIII for most patients. All plasma-derived products carry risks of transmitting hepatitis, HIV, parvovirus B19, transfusion-transmitted virus (TTV), and prion-induced (new variant Creutzfeldt-Jacob disease [nvCJD]) illnesses (see Complications and Hemophilia A for more information). A recombinant factor XIII (rFXIII) subunit A concentrate is available for use in congenital FXIII A-subunit deficiency. [99]
Development of FXIII inhibitors (alloantibodies or autoantibodies) is associated with significant morbidity and mortality.
Patient Education
Encourage patients to register with the local chapter of the National Hemophilia Foundation and to attend educational seminars. Provide one-on-one discussions of issues with patients and family members. Early and complete genetic testing can help families plan future pregnancies.
For patient education information, see What to Know about Coagulation Defects and the National Hemophilia Foundation's Factor XIII webpage.
-
Coagulation reactions leading to thrombin generation and activation of factor XIII.
-
Final steps in clot formation (from article: Factor XIII).
-
Activation of factor XIII and generation of insoluble cross-linked fibrin. Adapted from Lorand L. Ann N Y Acad Sci. 2001;936:291-311.
-
Postulated interaction between factor XIII and thrombin-activatable fibrinolytic inhibitor.
-
Cell surfaced–directed hemostasis. Initially, a small amount of thrombin is generated on the surface of the tissue factor–bearing (TF-bearing) cell. Following amplification, the second burst generates a larger amount of thrombin, leading to fibrin (clot) formation (from article: Factor XIII). Adapted from Hoffman and Monroe. Thromb Haemost. 2001;85(6):958-65.
-
Gene, messenger RNA, and protein for subunit A of factor XIII. Adapted from Reitsma PH. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Lippincott Williams & Wilkins; 2001:59-87 and from Roberts HR, Monroe DM III, Hoffman M. In: Williams Hematology. McGraw-Hill Professional; 2001:1409-34.
Tables
Properties |
A Chain |
B Chain |
Plasma FXIII |
Has 2 A chains |
Has 2 B chains |
Plasma level |
Approximately 15 mg/mL |
Approximately 21 mg/mL |
Chains are free in plasma |
No. All bound to B chain and present as an A2 B2 tetramer |
Yes. Excess B chain present in plasma as a B2 dimer |
Chain contains the catalytic site |
Yes |
No |
Chain is the carrier protein |
No |
Yes |
Chain acts as a brake on FXIII activation |
No |
Yes |
Cellular FXIII |
Has 2 A chains (A2 dimer) |
Has no B chains |
Mutations can lead to decreased FXIII activity |
Yes |
Yes |