Practice Essentials
The bone marrow failure syndromes comprise a group of disorders than can be either inherited or acquired. These diseases are intrinsic disorders of the bone marrow involving disruption in the homeostasis and function of hematopoietic stem cells, resulting in inadequate production of either a single or multiple cell lines (erythroid for red cells, myeloid for white blood cells, megakaryocytic for platelets). The lymphocytes, which are involved in lymphoproliferative disorders, are usually spared (see the image below). (See Etiology.)
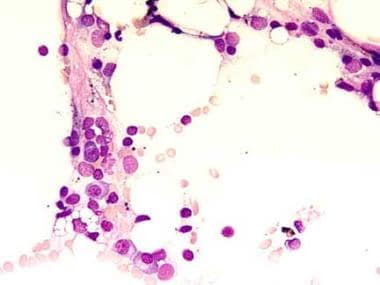 This bone marrow film at 400X magnification demonstrates a complete absence of hemopoietic cells. Most of the identifiable cells are lymphocytes or plasma cells. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland (http://www.aum.iawf.unibe.ch/).
This bone marrow film at 400X magnification demonstrates a complete absence of hemopoietic cells. Most of the identifiable cells are lymphocytes or plasma cells. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland (http://www.aum.iawf.unibe.ch/).
The inherited bone marrow failure syndromes (IBMFS) include Fanconi anemia, dyskeratosis congenita, Diamond-Blackfan anemia, and other genetic disorders. [1] The most common cause of acquired bone marrow failure is aplastic anemia. [2] (See Etiology, Presentation, Workup, and Treatment.)
Diseases that can present in a manner similar to acquired bone marrow failure include myelodysplastic syndromes, paroxysmal nocturnal hemoglobinuria, and large granular lymphocytic leukemia. (See DDx.)
For patient education information, see Anemia.
Etiology
Bone marrow failure can be either inherited or acquired and can involve a single hematopoietic stem cell line or all three cell lines. These etiologies involve the following:
- A decrease in or damage to the hematopoietic stem cells and their microenvironment, resulting in hypoplastic or aplastic bone marrow
- Maturation defects, such as in vitamin B12 or folate deficiency
- Differentiation defects, such as myelodysplasia
Damage to hematopoietic stem cells can be congenital or acquired. Mechanisms include the following:
-
An acquired stem cell injury from viruses, toxins, or chemicals (eg, chloramphenicol, insecticides [3] ) that leads to a quantitative or qualitative abnormality
-
Abnormal humoral or cellular control of hematopoiesis
-
An abnormal or hostile marrow microenvironment
-
Immunologic suppression of hematopoiesis (ie, mediated by antibodies, T cells [or cellularly], or lymphokines)
-
Mutations in genes, causing inherited bone marrow failure syndromes; identification of these relevant mutations has led to progress in defining the precise functions of the corresponding proteins in normal cells
Inherited bone marrow failure syndromes
The genetic abnormalities in the inherited bone marrow failure syndromes (IBMFS) have been identified in the following disorders [4, 5] :
-
Dyskeratosis congenita
-
Shwachman-Diamond syndrome
-
Diamond-Blackfan anemia
-
Amegakaryocytic thrombocytopenia
-
Congenital neutropenia
-
Telomere biology disorders
-
GATA2 deficiency syndrome
-
SAMD9/SAMD9L syndromes
-
Thrombocytopenia syndromes
Fanconi anemia is inherited in either an autosomal recessive or X-linked fashion. Twelve Fanconi anemia complementation (FANC) group genes have been identified. These genes collaborate in a complicated pathway (FA pathway) that is responsible for the repair of DNA damage. One of these genes (FANCD1) is the breast/ovarian susceptibility gene (BRCA2).
Dyskeratosis congenita is inherited in an X-linked recessive, autosomal dominant, or autosomal recessive manner. Patients with the X-linked form have mutations in DKC1 at band Xq28, a gene that encodes for dyskenin, in a protein involved in the telomere maintenance pathway. Other patients have mutations in band 3q26 in TERC, a part of the telomerase complex, and still others have mutations in the telomerase reverse transcription (TERT) enzyme. [6]
Shwachman-Diamond syndrome is an autosomal recessive disorder in which the majority of patients have a mutation in the Shwachman Bodian Diamond syndrome gene (SBDS), located at band 7q11.
Amegakaryocytic thrombocytopenia is an autosomal recessive disorder with biallelic mutations in the thrombopoietin receptor, MPL, at the band 1p34 location.
Diamond-Blackfan anemia is an autosomal dominant disease in which 25% of patients have a mutation in the gene for small ribosomal protein (RPS19), located at band 19q13.2.
In half of the patients, severe congenital neutropenia is associated with dominant mutations in neutrophil elastase (ELA2, located at band 19p13.3), while a few patients have mutations in GFI-1.
Thrombocytopenia-absent radii (TAR) syndrome is associated with bone marrow failure. Identification of a heterozygous null allele (most often a minimally deleted 200-kb region at chromosome band 1q21.1) in trans with a heterozygous RBM8A hypomorphic allele on molecular genetic testing confirms the diagnosis of TAR syndrome. [7]
Germline mutations in GATA2 cause an autosomal dominant heterogeneous IBMFS characterized by susceptibility to infection, pulmonary and vascular/lymphatic dysfunction, autoimmunity, and malignancy. Wlodarski and colleagues identified germline GATA2 mutations in 28 (7%) of 426 children age 18 years or younger with sporadic MDS in Germany. [8]
Next-generation sequencing has broadened the spectrum of possible etologic germline mutations. In a cohort of 179 patients (from 173 families) with bone marrow failure of suspected inherited origin, genomic DNA from skin fibroblasts using whole-exome sequencing were analyzed. Causal or likely causal germ line mutations were assigned in 86 patients (48.0%), involving a total of 28 genes. These included genes in familial hematopoietic disorders (GATA2, RUNX1), telomeropathies (TERC, TERT, RTEL1), ribosome disorders (SBDS, DNAJC21, RPL5), and DNA repair deficiency (LIG4). [9]
Many patients had an atypical presentation, and the mutated gene was often not clinically suspected. Mutations in genes seldom reported in IBMFS were also identified, such as SAMD9 and SAMD9L (N = 16 of the 86 patients, 18.6%), MECOM/EVI1 (N = 6, 7.0%), and ERCC6L2 (N = 7, 8.1%), each of which was associated with a distinct natural history; SAMD9 and SAMD9L patients often experienced transient aplasia and monosomy 7, whereas MECOM patients presented early-onset severe aplastic anemia, and ERCC6L2 patients, mild pancytopenia with myelodysplasia. [9]
Constitutional causes
Constitutional aplastic anemia is associated with chronic bone marrow failure, congenital anomalies, familial incidence, or thrombocytopenia at birth. Constitutional causes of aplastic anemia include the following conditions:
-
Fanconi anemia - Characterized by familial aplastic anemia, chromosomal breaks, and, in some cases, congenital anomalies of the thumb or kidneys
-
Dyskeratosis congenita - Another rare disorder, dyskeratosis congenita has a characteristic dermatologic manifestation of nail dystrophies and leukoplakia; patients with this disease develop aplastic anemia in their second decade of life
-
Shwachman-Diamond syndrome - This disorder consists of exocrine pancreatic insufficiency and bone marrow failure occasionally accompanied by cartilage and hair hypoplasia resulting in short stature and dysostosis
Single cytopenias
Pure red cell aplasia can be secondary to thymoma, collagen vascular diseases (eg, systemic lupus erythematosus) or pregnancy. It can occur transiently resulting from a viral infection, as with parvovirus B19. Pure red cell aplasia can also be permanent, as a result of viral hepatitis. Finally, it can arise from lymphoproliferative diseases (eg, lymphomas, chronic lymphocytic leukemia).
Amegakaryocytic thrombocytopenic purpura has been reported to occur as a result of causes similar to those for pure red cell aplasia.
Early forms of myelodysplastic syndrome initially can manifest as a single cytopenia or, more often, as a bicytopenias.
Pancytopenia
A decrease in all three cell lines is the most common manifestation of bone marrow failure. Aplastic or hypoplastic anemia can be idiopathic in nature, or it can develop from secondary causes. Myelodysplastic anemia also can cause pancytopenia. Myelophthisic anemia may result from marrow destruction because of tumor invasion or granulomas.
Epidemiology
The incidence of bone marrow failure resulting from hypoplastic or aplastic anemia is low in the United States and Europe (2-6 cases per million persons) compared with that of bone marrow failure resulting from acute myelogenous leukemia and multiple myeloma (27-35 cases per million persons). The frequency of myelodysplasia, on the other hand, has increased from 143 cases reported in 1973 to about 15,000 cases annually in United States. This is an underestimation of the actual incidence, which is believed to be about 35,000-55,000 new cases a year.
The frequency of bone marrow failure is at least 3 times higher in East Asia than it is in the United States and Europe. Mexico and Latin America also have high occurrence rates of bone marrow failure, attributed to the liberal use of chloramphenicol. In addition, environmental factors and the pervasive use of insecticides have been implicated as causes of this disease. The incidence of myelodysplasia has been estimated to be around 4-5 per 100,000 population per year in Germany and Sweden.
Prognosis
The prognosis of bone marrow failure depends on the duration of the marrow function abnormality. Most inherited forms of bone marrow failure, such as Fanconi anemia, are associated with transformation into leukemia several years later. Transient causes of bone marrow failure, such as parvovirus infections, are usually self-limiting.
Acquired idiopathic aplastic anemia is usually permanent and life threatening with 6-month mortality of 50% from initial diagnosis.
Morbidity and mortality
Bone marrow failure resulting in failure to produce one, two, or all three blood cell lines increases patient morbidity and mortality.
Morbidity and mortality from pancytopenia are caused by low levels of mature blood cells. Severe anemia can cause high-output cardiac failure and fatigue. Neutropenia can predispose individuals to bacterial and fungal infections. Thrombocytopenia can cause spontaneous bleeding and hemorrhage.
The severity and extent of the cytopenia(s) determines prognosis. Severe pancytopenia is a medical emergency, requiring rapid institution of definitive therapy (ie, early determination of supportive care and bone marrow transplant candidates).
Transfusion complications
Over time, the transfusion of packed red cells increases the patient’s total iron load. Increased levels of iron are toxic to various organs, including the heart and can result in arrhythmias by blocking the bundle of His, diabetes by damaging the islets of Langerhans in the pancreas, and liver cirrhosis. (Iron can also produce bronze coloration in fair-skinned individuals.) Therefore, it is necessary to measure a patient’s iron stores (in the form of ferritin).
Administering a chelating agent is an effective method of removing excess iron. Chelating agents are composed of molecules that bind tightly with free iron and remove the iron by carrying it as the agents are excreted from the body.
Deferoxamine is the iron chelator available in parenteral form. If given intravenously, its activity is short and it is excreted rapidly by the kidneys. A subcutaneous infusion given continuously by a portable pump for 3-4 hours every 12 hours is the preferred method. It optimizes the binding of the chelator to the free iron. As more free iron is excreted, storage iron is mobilized into the free form. This treatment can be performed in an outpatient setting. Deferiprone and deferasirox are two oral iron chelating agents that can be used alone or in conjunction with deferoxamine.
Monitoring serum ferritin levels and measuring total iron urinary excretion can determine the effectiveness of therapy. Most tissue damage can be reversed with timely chelation, except for cirrhosis of the liver (once it has set in).
-
This bone marrow film at 400X magnification demonstrates a complete absence of hemopoietic cells. Most of the identifiable cells are lymphocytes or plasma cells. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland (http://www.aum.iawf.unibe.ch/).