Background
Although still in use clinically, it is important to note that systemic inflammatory response syndrome(SIRS) as a definition has been abandoned since 2016. This has occurred mainly as mortality prediction using SIRS alone is poor compared with Sequential Organ Failure Assessment (SOFA). Furthermore, sepsis has been redefined as requiring organ dysfunction. [1]
In 2016, the Society of Critical Care Medicine (SCCM) and the European Society of Intensive Care Medicine (ESICM) led a task force to propose a new definition for sepsis. Sepsis-3 was born, defined as "life-threatening organ dysfunction caused by a dysregulated host response to infection." [1, 2] This created a paradigm shift in the sepsis landscape. Although created in 1998, [3] and largely not designed to predict mortality like Acute Physiology and Chronic Health Evaluation (APACHE) II or Simplified Acute Physiology Score (SAPS), the nondiagnostic intensive care unit (ICU) mortality prediction score SOFA was recommended by the task force owing to SOFA's superior predictive validity for in-hospital mortality as compared with SIRS. In other words, SOFA was endorsed by the 2016 SCCM/ESICM Task Force to identify in-hospital patients at risk of death due to sepsis. The following system assessments comprise the SOFA severity score:
-
Respiratory system - The ratio of arterial oxygen tension to fraction of inspired oxygen (PaO 2/FiO 2)
-
Cardiovascular system - Mean arterial pressure (MAP) and presence of vasopressor medication(s)
-
Hepatic system - Bilirubin level
-
Coagulation system - Platelet concentration
-
Neurologic system - Glasgow Coma Score
-
Renal system - Serum creatinine or urine output
SOFA as a clinical tool can be accessed here: SOFA Calculator.
In 2001, the SCCM/ESICM, along with other leading organizations, including the American College of Chest Physicians (CHEST), the American Thoracic Society (ATS), and the Surgical Infection Society (SIS), convened as the Consensus Conference reviewing the 1992 SIRS criteria to establish the general definitions known as Sepsis-2, leaving the definitions of sepsis and septic shock unchanged for the next twenty years. [4]
In 1992, CHEST and the Society of Critical Care Medicine (SCCM) introduced definitions for SIRS, sepsis, severe sepsis, septic shock, and multiple organ dysfunction syndrome (MODS). [5] The idea behind defining SIRS was to define a clinical response to a nonspecific insult of either infectious or noninfectious origin. SIRS is defined as two or more of the following variables (see Presentation and Workup):
-
Fever of more than 38°C (100.4°F) or less than 36°C (96.8°F)
-
Heart rate of more than 90 beats per minute
-
Respiratory rate of more than 20 breaths per minute or arterial carbon dioxide tension (PaCO 2) of less than 32 mm Hg
-
Abnormal white blood cell count (>12,000/µL or < 4,000/µL or >10% immature [band] forms)
SIRS is nonspecific and can be caused by ischemia, inflammation, trauma, infection, or several insults combined. Thus, SIRS is not always related to infection. Although sepsis has diverged from SIRS criteria for diagnosis and management in recent years, focusing more on infectious etiologies, the pathophysiologic processes present in sepsis and noninfectious SIRS are remarkably similar, making a discussion of SIRS in critical illness appropriate.(See Pathophysiology and Etiology.)
See the diagram below.
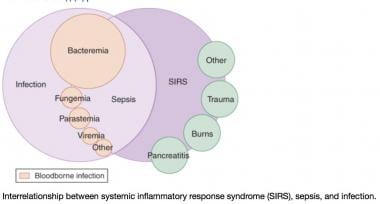 A Venn diagram of the systemic inflammatory response syndrome (SIRS). The diagram shows the overlap between infection, bacteremia, sepsis, SIRS, and multiorgan dysfunction. This perspective shows that not only infection can cause SIRS. Courtesy of emDocs.net (http://www.emdocs.net/mimics-of-sepsis/).
A Venn diagram of the systemic inflammatory response syndrome (SIRS). The diagram shows the overlap between infection, bacteremia, sepsis, SIRS, and multiorgan dysfunction. This perspective shows that not only infection can cause SIRS. Courtesy of emDocs.net (http://www.emdocs.net/mimics-of-sepsis/).
Bacteremia, sepsis, and septic shock
Infection is defined as "a microbial phenomenon characterized by an inflammatory response to the microorganisms or the invasion of normally sterile tissue by those organisms."
Bacteremia is the presence of bacteria within the bloodstream, but this condition does not always lead to SIRS or sepsis. Sepsis is the systemic response to infection and is defined as the presence of SIRS in addition to a documented or presumed infection. Severe sepsis meets the aforementioned criteria and is associated with organ dysfunction, hypoperfusion, or hypotension. (See Etiology, Treatment, and Medication.)
Sepsis-induced hypotension is defined as "the presence of a systolic blood pressure of less than 90 mm Hg or a reduction of more than 40 mm Hg from baseline in the absence of other causes of hypotension." Patients meet the criteria for septic shock if they have persistent hypotension and perfusion abnormalities despite adequate fluid resuscitation. MODS is a state of physiologic derangements in which organ function is not capable of maintaining homeostasis. (See Pathophysiology.)
Although not universally accepted terminology, severe SIRS and SIRS shock are terms that some authors have proposed. These terms suggest organ dysfunction or refractory hypotension related to an ischemic or inflammatory process rather than to an infectious etiology.
Pathophysiology
Systemic inflammatory response syndrome (SIRS), independent of the etiology, has the same pathophysiologic properties, with minor differences in inciting cascades. Many consider the syndrome a self-defense mechanism. Inflammation is the body's response to nonspecific insults that arise from chemical, traumatic, or infectious stimuli. The inflammatory cascade is a complex process that involves humoral and cellular responses, complement, and cytokine cascades. Bone [5] best summarized the relationship between these complex interactions and SIRS as the following 3-stage process.
Stage I
Following an insult, cytokines are produced within immune effector cells de novo at the site. Local cytokine production incites a cellular inflammatory response, thereby promoting wound repair and recruitment of the reticular endothelial system. This process is essential for normal host defense homeostasis and if absent is not compatible with life. Local inflammation, such as in the skin and subcutaneous soft tissues, carries the classic description of rubor, tumor, dolor, calor and functio laesa.
Rubor or redness reflects local vasodilation caused by release of local vasodilating substances like nitric oxide (NO) and prostacyclin.
Tumor or swelling is due to vascular endothelial tight junction disruption and the local extravasation of protein-rich fluid into the interstitium, which also allows activated white blood cells to pass from the vascular space into the tissue space to help clear infection and promote repair.
Dolor is pain and represents the impact inflammatory mediators have on local somatosensory nerves. Presumably, this pain stops the host from trying to use this part of his or her body as it tries to repair itself.
Calor is the increased heat primarily due to increased blood flow but also increased local metabolism as white blood cells become activated and localize to the injured tissue.
Finally, functio laesa is loss of function, a hallmark of inflammation and a common clinical finding of organ dysfunction with the infection is isolated to a specific organ (eg, pneumonia—acute respiratory failure; kidney—acute kidney injury).
Importantly, on a local level, this cytokine and chemokine release by attracting activated leukocytes to the region may cause local tissue destruction (eg, abscess) or cellular injury (eg, pus), which appear to be the necessary byproducts of an effective local inflammatory response.
Stage II
Small quantities of local cytokines are released into the circulation, improving the local response. This leads to growth factor stimulation and the recruitment of macrophages and platelets. This acute phase response is typically well controlled by a decrease in the proinflammatory mediators and by the release of endogenous antagonists; the goal is homeostasis. At this stage, some minimal malaise and low-grade fever may become manifest.
Stage III
If homeostasis is not restored and if the inflammatory stimuli continue to seed into the systemic circulation, a significant systemic reaction occurs. The cytokine release leads to destruction rather than protection. A consequence of this is the activation of numerous humoral cascades and the activation of the reticular endothelial system and subsequent loss of circulatory integrity. This leads to end-organ dysfunction.
Multi-hit theory
Bone also endorsed a multi-hit theory behind the progression of SIRS to organ dysfunction and possibly multiple organ dysfunction syndrome (MODS). In this theory, the event that initiates the SIRS cascade primes the pump. With each additional event, an altered or exaggerated response occurs, leading to progressive illness. The key to preventing the multiple hits is adequate identification of the cause of SIRS and appropriate resuscitation and therapy.
Inflammatory cascade
Trauma, inflammation, or infection leads to the activation of the inflammatory cascade. Initially, a proinflammatory activation occurs, but almost immediately thereafter a reactive suppressing anti-inflammatory response occurs. This SIRS usually manifests itself as increased systemic expression of both proinflammatory and anti-inflammatory species. When SIRS is mediated by an infectious insult, the inflammatory cascade is often initiated by endotoxin or exotoxin. Tissue macrophages, monocytes, mast cells, platelets, and endothelial cells are able to produce a multitude of cytokines. The cytokines tissue necrosis factor–alpha (TNF-α) and interleukin-1 (IL-1) are released first and initiate several cascades.
The release of IL-1 and TNF-α (or the presence of endotoxin or exotoxin) leads to cleavage of the nuclear factor-kB (NF-kB) inhibitor. Once the inhibitor is removed, NF-kB is able to initiate the production of messenger ribonucleic acid (mRNA), which induces the production other proinflammatory cytokines.
IL-6, IL-8, and interferon gamma are the primary proinflammatory mediators induced by NF-kB. In vitro research suggests that glucocorticoids may function by inhibiting NF-kB. TNF-α and IL-1 have been shown to be released in large quantities within 1 hour of an insult and have both local and systemic effects. In vitro studies have shown that these 2 cytokines given individually produce no significant hemodynamic response but that they cause severe lung injury and hypotension when given together. TNF-α and IL-1 are responsible for fever and the release of stress hormones (norepinephrine, vasopressin, activation of the renin-angiotensin-aldosterone system).
Other cytokines, especially IL-6, stimulate the release of acute-phase reactants such as C-reactive protein (CRP) and procalcitonin. Of note, infection has been shown to induce a greater release of TNF-α —thus inducing a greater release of IL-6 and IL-8—than trauma does. This is suggested to be the reason higher fever is associated with infection rather than trauma.
The proinflammatory interleukins either function directly on tissue or work via secondary mediators to activate the coagulation cascade and the complement cascade and the release of nitric oxide, platelet-activating factor, prostaglandins, and leukotrienes.
High mobility group box 1 (HMGB1) is a protein present in the cytoplasm and nuclei in a majority of cell types. In response to infection or injury, as is seen with SIRS, HMGB1 is secreted by innate immune cells and/or released passively by damaged cells. Thus, elevated serum and tissue levels of HMGB1 would result from many of the causes of SIRS.
HMGB1 acts as a potent proinflammatory cytokine and is involved in delayed endotoxin lethality and sepsis. [6] In an observational study of patients with traumatic brain injury, multivariate analysis selected plasma HMGB1 level as an independent predictor for 1-year mortality and unfavorable outcome. [7] Therapeutic studies are under way to evaluate various mechanisms to block HMGB1, with hopes of improving outcomes in SIRS and sepsis syndromes. [6]
Numerous proinflammatory polypeptides are found within the complement cascade. Protein complements C3a and C5a have been the most studied and are felt to contribute directly to the release of additional cytokines and to cause vasodilatation and increasing vascular permeability. Prostaglandins and leukotrienes incite endothelial damage, leading to multiorgan failure.
Polymorphonuclear cells (PMNs) from critically ill patients with SIRS have been shown to be more resistant to activation than PMNs from healthy donors, but, when stimulated, demonstrate an exaggerated microbicidal response. This may represent an autoprotective mechanism in which the PMNs in the already inflamed host may avoid excessive inflammation, thus reducing the risk of further host cell injury and death. [8]
Coagulation
The correlation between inflammation and coagulation is critical to understanding the potential progression of SIRS. IL-1 and TNF-α directly affect endothelial surfaces, leading to the expression of tissue factor. Tissue factor initiates the production of thrombin, thereby promoting coagulation, and is a proinflammatory mediator itself. Fibrinolysis is impaired by IL-1 and TNF-α via production of plasminogen activator inhibitor-1. Proinflammatory cytokines also disrupt the naturally occurring anti-inflammatory mediators antithrombin and activated protein-C (APC).
If unchecked, this coagulation cascade leads to complications of microvascular thrombosis, including organ dysfunction. The complement system also plays a role in the coagulation cascade. Infection-related procoagulant activity is generally more severe than that produced by trauma.
SIRS versus CARS
The cumulative effect of this inflammatory cascade is an unbalanced state with inflammation and coagulation dominating. To counteract the acute inflammatory response, the body is equipped to reverse this process via the counter-inflammatory response syndrome (CARS). IL-4 and IL-10 are cytokines responsible for decreasing the production of TNF-α, IL-1, IL-6, and IL-8. In fact, this proinflammatory and anti-inflammatory activation mirrors other homeostatic processes, like coagulation, anticoagulation, complement activation, and complement suppression.
Clearly, the normal homeostatic processes attempt to keep these very toxic inflammatory processes in check. Inflammation is an essential component of host defense and serves a very strongly positive survival function in suppressing and then eliminating local infection and tissue injury. It is only when this localized aggressive injury process gains access to the whole body through the blood stream and lymphatics that a SIRS develops.
The acute phase response also produces antagonists to TNF-α and IL-1 receptors. These antagonists either bind the cytokine, and thereby inactivate it, or block the receptors. Comorbidities and other factors can influence a patient's ability to respond appropriately.
The balance of SIRS and CARS helps determine a patient's outcome after an insult. Some researchers believe that because of CARS, medications meant to inhibit the proinflammatory mediators may lead to deleterious immunosuppression.
Etiology
The etiology of systemic inflammatory response syndrome (SIRS) is broad and includes infectious and noninfectious conditions, surgical procedures, trauma, medications, and therapies. The inciting molecular stimuli inducing the above generalized inflammatory reaction fall into two broad categories, pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PAMPs become present when infection of foreign cell lysis releases these foreign molecules intrinsic to their structure into the circulation, whereas DAMPs arise when cellular injury occurs at rates that overwhelm local clearance mechanisms. Thus, it can be seen that generalized bacteremia, severe pneumonia (viral or bacterial), severe trauma with tissue injury, and pancreatitis all share common inflammatory activation pathways.
The following is partial list of the infectious causes of SIRS:
-
Bacterial sepsis
-
Burn wound infections
-
Candidiasis
-
Cellulitis
-
Cholecystitis
-
Community-acquired pneumonia [9]
-
Diabetic foot infection
-
Erysipelas
-
Infective endocarditis
-
Influenza
-
Intra-abdominal infections (eg, diverticulitis, appendicitis)
-
Gas gangrene
-
Meningitis
-
Nosocomial pneumonia
-
Pseudomembranous colitis
-
Pyelonephritis
-
Septic arthritis
-
Urinary tract infections (male and female)
The following is a partial list of the noninfectious causes of SIRS:
-
Acute mesenteric ischemia
-
Adrenal insufficiency
-
Autoimmune disorders
-
Burns
-
Chemical aspiration
-
Cirrhosis
-
Cutaneous vasculitis
-
Dehydration
-
Drug reaction
-
Electrical injuries
-
Erythema multiforme
-
Hemorrhagic shock
-
Hematologic malignancy
-
Intestinal perforation
-
Medication side effect (eg, from theophylline)
-
Myocardial infarction
-
Pancreatitis [10]
-
Seizure
-
Substance abuse - Stimulants such as cocaine and amphetamines
-
Surgical procedures
-
Toxic epidermal necrolysis
-
Transfusion reactions
-
Upper gastrointestinal bleeding
-
Vasculitis
Epidemiology
Occurrence in the United States
The true incidence of systemic inflammatory response syndrome (SIRS) is unknown but probably very high, owing to the nonspecific nature of its definition. Not all patients with SIRS require hospitalization or have diseases that progress to serious illness. Indeed, patients with a seasonal head cold due to rhinovirus usually fulfill the criteria for SIRS. Because SIRS criteria are nonspecific and occur in patients who present with conditions ranging from influenza to cardiovascular collapse associated with severe pancreatitis, [10] any incidence figures would need to be stratified based on SIRS severity.
Rangel-Fausto et al published a prospective survey of patients admitted to a tertiary care center that revealed 68% of hospital admissions to surveyed units met SIRS criteria. [11] The incidence of SIRS increased as the level of unit acuity increased. The following progression of patients with SIRS was noted: 26% developed sepsis, 18% developed severe sepsis, and 4% developed septic shock within 28 days of admission.
Pittet et al performed a hospital survey of SIRS that revealed an overall in-hospital incidence of 542 episodes per 1000 hospital days. [12] In comparison, the incidence in the intensive care unit (ICU) was 840 episodes per 1000 hospital days. It is not clear what percentage of patients with SIRS have a primary infectious etiology, allowing them to be classified as having sepsis. However, most likely the proportion of SIRS patients varies across patient and hospital groups, being highest for example in acute care settings and in those with immune deficiency.
The etiology of patients admitted with severe sepsis from a community emergency department was evaluated by Heffner et al, who determined that 55% of patients had negative cultures and that 18% were diagnosed with noninfectious causes that mimicked sepsis (SIRS). Many of the noninfectious etiologies required urgent alternate disease-specific therapy (eg, pulmonary embolism, myocardial infarction, pancreatitis). Of the SIRS patients without infection, the clinical characteristics were similar to those with positive cultures. [13]
Another study demonstrated that 62% of patients who presented to the emergency department with SIRS had a confirmed infection, while 38% did not. Within the same cohort of patients, 38% of infected patients did not present with SIRS. [14]
Still, Angus et al found the incidence of severe SIRS associated with infection to be 3 cases per 1,000 population, or 2.26 cases per 100 hospital discharges. [15] The real incidence of SIRS, therefore, must be much higher and likely depends somewhat on the rigor with which the definition is applied.
International occurrence
No difference in the frequency of SIRS exists based on world geography.
Sex-related demographics
The sex-based mortality risk of severe SIRS is unknown. Females tend to have less inflammation from the same degree of proinflammatory stimuli because of the mitigating aspects of estrogen. The reasons for this are not completely known, but estrogen sustains adrenergic receptor activity in inflammation, when, in its absence, adrenergic receptor down-regulation occurs. Thus, premenopausal females tend to have less vasoplegia and respond more vigorously to resuscitation efforts. This equates to women having a 10-year age benefit over men. The mortality rate among women with severe sepsis is similar to that of men who are 10 years younger; however, whether this protective effect applies to women with noninfectious SIRS is unknown.
Age-related demographics
Extremes of age (young and old) and concomitant comorbidities probably negatively affect the outcome of SIRS. Young people may be able to mount a more exuberant inflammatory response to a challenge than older people and yet may be able to better modify the inflammatory state (via the counter-inflammatory response syndrome [CARS]). Young people have better outcomes for equivalent diagnoses.
Prognosis
Comstedt et al, in a study of systemic inflammatory response syndrome (SIRS) in acutely hospitalized medical patients, demonstrated a 6.9 times higher 28-day mortality in SIRS patients than in non-SIRS patients. Most deaths occurred in SIRS patients with an associated malignancy. [14]
Prognosis depends on the etiologic source of SIRS, as well as on associated comorbidities. The mortality rates in the previously mentioned Rangel-Fausto et al study were 7% (SIRS), 16% (sepsis), 20% (severe sepsis), and 46% (septic shock). [11] The median time interval from SIRS to sepsis was inversely related to the number of SIRS criteria met. Morbidity is related to the causes of SIRS, complications of organ failure, and the potential for prolonged hospitalization.
However, the large retrospective study of all of Australia and New Zealand ICU care from 2000-2012 demonstrated a clear progressive decline in severe sepsis and septic shock mortality from 35% to 18% over this period, with equal trends across all age groups and treatment settings. [16] These data suggest that attention to detail, using best practices and overall quality care, has nearly halved mortality from severe sepsis independent of any specific treatment. Thus, attention to overall patient status and use of proven risk reduction approaches (eg, stress ulcer prophylaxis, DVT prophylaxis, daily awakening, and weaning trials in ventilator-dependent patients) are central to improving outcome from severe sepsis.
Pittet et al showed that control patients had the shortest hospital stay, while patients with SIRS, sepsis, and severe sepsis, respectively, required progressively longer hospital stays. [12]
A study by Shapiro et al evaluated mortality in patients with suspected infection in the emergency department and found the following in-hospital mortality rates [17] :
-
Suspected infection without SIRS - 2.1%
-
Sepsis - 1.3%
-
Severe sepsis - 9.2%
-
Septic shock - 28%
In the study, the presence of SIRS criteria alone had no prognostic value for either in-hospital mortality or 1-year mortality. Each additional organ dysfunction increased the risk of mortality at 1 year. The authors concluded that organ dysfunction, rather than SIRS criteria, was a better predictor of mortality.
Sinning et al evaluated the SIRS criteria in patients who underwent transcatheter aortic valve implantation (TAVI) and found that SIRS appeared to be a strong predictor of mortality. The occurrence of SIRS was characterized by a significantly elevated release of IL-6 and IL-8, with subsequent increase in the leukocyte count, C-reactive protein (CRP), and procalcitonin. The occurrence of SIRS was related to 30-day and 1-year mortality (18% vs 1.1% and 52.5% vs 9.9%, respectively) and independently predicted 1-year mortality risk. [18]
In the aforementioned Heffner et al study, patients without an identified infection had a lower hospital mortality rate than did patients with an infectious etiology for their SIRS (9% vs 15%, respectively). [13]
As mentioned in Background, the redefined definitions by the 2016 SCCM/EISCM task force support that (1) sepsis requires organ dysfunction and (2) the concepts of SIRS and severe sepsis are eliminated. Furthermore, the task force recommends SOFA as the tool to assess severity of organ dysfunction in a potentially septic patient; the authors had concluded that SOFA has superior predictive validity for in-hospital mortality as compared with SIRS. [2]
Patient Education
Education should ideally target the patient's family. Family members need to understand the fluid nature of immune responsiveness and that SIRS is a potential harbinger of other more dire syndromes.
-
A Venn diagram of the systemic inflammatory response syndrome (SIRS). The diagram shows the overlap between infection, bacteremia, sepsis, SIRS, and multiorgan dysfunction. This perspective shows that not only infection can cause SIRS. Courtesy of emDocs.net (http://www.emdocs.net/mimics-of-sepsis/).