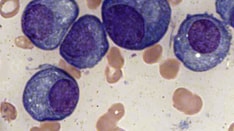
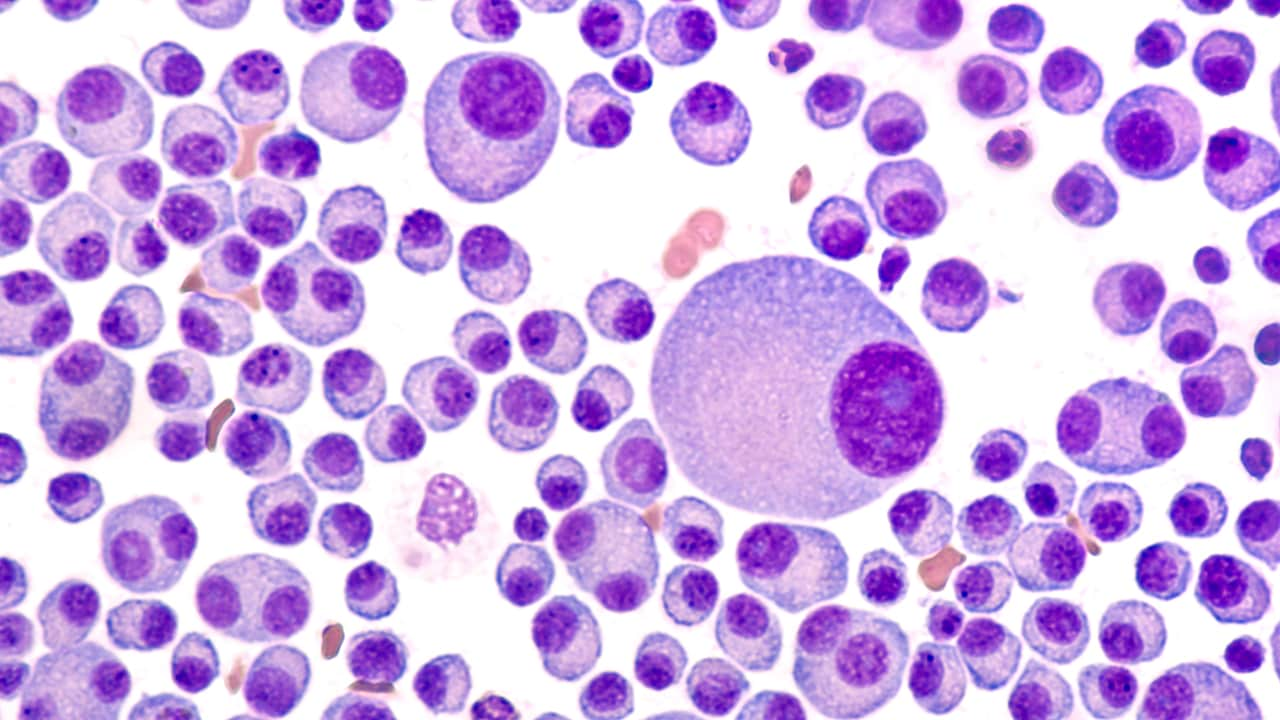
Practice Essentials
Light chains (molecular weight 22,000 d) are polypeptides that are synthesized by plasma cells and form part of immunoglobulins. Plasma cells normally produce a slight excess of light chains that are either excreted or catabolized by the kidney, and only a minute amount of light-chain protein normally appears in the urine. Light-chain proteins appear in urine in high concentration either when the production of light-chain proteins is markedly increased or when the ability of the proximal tubules to reabsorb all the filtered protein is diminished.
The most characteristic histologic lesion of light chain deposition disease (LCDD) is nodular glomerulosclerosis, which must be distinguished from diabetic glomerulosclerosis by using electron microscopy. [1] See the image below.
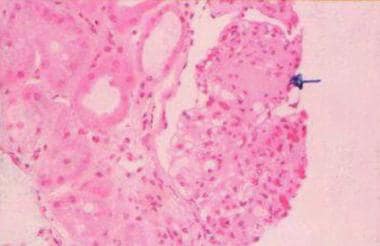 Light chain–associated renal disorders. Light microscopy (hematoxylin and eosin stain at 25X power) showing nodular glomerulosclerosis (arrow) and thickening of the basement membrane. Courtesy of Madeleine Moussa, MD, FRCPC, Department of Pathology, London Health Sciences Centre, London, Ontario, Canada.
Light chain–associated renal disorders. Light microscopy (hematoxylin and eosin stain at 25X power) showing nodular glomerulosclerosis (arrow) and thickening of the basement membrane. Courtesy of Madeleine Moussa, MD, FRCPC, Department of Pathology, London Health Sciences Centre, London, Ontario, Canada.
The term Bence Jones protein has been used to designate a urinary protein that leaves solution at approximately 56°C under certain conditions of pH and ionic strength and returns to the solution upon further heating to 100°C. The Bence Jones protein represents a homogeneous population of immunoglobulin light chains of either kappa type or lambda type and is the product of a presumed single clone of plasma cells. The presence of light-chain proteins in the urine is associated with a number of systemic diseases (see Etiology).
Smithline et al first used the term light-chain nephropathy in 1976 to describe a case of renal tubular dysfunction with light-chain proteinuria. [2] The term has been associated with various glomerular abnormalities that are caused by the deposition of these monoclonal immunoglobulins (or their heavy-chain or light-chain subunits) and are broadly classified into 2 categories, organized or nonorganized, depending on the pattern of deposition.
Organized deposits include the following:
-
Fibrillar (amyloidosis)
-
Microtubular (cryoglobulinemia, immunotactoid glomerulonephritis)
Nonorganized, granular deposits include the following:
-
Monoclonal immunoglobulin deposition disease (MIDD)
-
Light-chain, heavy-chain, and light- and heavy-chain deposition disease
Pathophysiology
Normal (renal) handling of light-chain proteins
Light chains (molecular weight 22,000 d) are polypeptides synthesized by plasma cells and assembled with heavy chains to form the various classes of immunoglobulins, for example, immunoglobulin G (IgG), immunoglobulin M (IgM), and immunoglobulin A (IgA). Plasma cells normally produce a slight excess of light chains that are either excreted or catabolized by the kidney.
Light chains are divided into 2 major classes based on the amino acid sequence in the constant portion of the polypeptide chain and are designated as kappa and lambda. These are further divided into at least 10 subtypes (4 kappa and 6 lambda) based on the amino acid sequence in the variable region of the polypeptide chain. Individual immunoglobulins have either kappa or lambda light chains, but not both.
Kappa light chains usually exist as monomers (22,000 d) and are therefore small enough to be filtered through the glomerulus, but they may exist as dimers. Lambda light chains usually exist as dimers (44,000 d) and, therefore, are less likely to be filtered and appear in urine. At times, light chains of either kappa or lambda type may form tetramers (88,000 d), which are not filtered, and a patient may have light-chain proteinemia without light-chain proteinuria.
The kidney is the major site of metabolism of light-chain proteins. The filtered light-chain proteins, reabsorbed by the proximal tubular cells via the tandem megalin/cubilin receptors, are catabolized by lysosomal enzymes. This process is exceedingly efficient, and only a minute amount of light-chain protein normally appears in the urine.
Metabolism (catabolism) of these filtered light-chain proteins depends on normal proximal tubular cell function, and damage to these cells can result in increased excretion of light-chain proteins in the urine. Hence, light-chain proteins appear in urine in high concentration either when the production of light-chain proteins is markedly increased or when the ability of the proximal tubules to reabsorb all the filtered protein is exceeded.
Glomerulopathic light-chains (G-LC) interact with mesangial cells and alter the mesangial homeostasis in 2 different ways, depending on whether G-LC is from a patient with LCCDD or amyloidosis. In contrast, the tubulopathic light chains (T-LC) from patients with myeloma cast nephropathy do not significantly interact with mesangial cells and do not alter mesangial homeostasis. Some of these light chains are toxic to proximal tubule cells and induce inflammatory/proinflammatory cytokines that may contribute to kidney disease in myeloma.
Light-chain proteins may manifest in the urine because of the following:
-
Asymptomatic light-chain proteinuria
-
Proximal tubular dysfunction (ie, Fanconi syndrome)
-
Light chain deposition disease (ie, nodular glomerulosclerosis or, rarely, glomerulonephritis)
-
Cast nephropathy
-
Amyloidosis
The isoelectric point (pI) of the light chain may be an important determinant of its potential for inducing renal damage. Proteins with a relatively high pI (> 5.8-6) appear to be more likely to be associated with renal failure. These light chains have a cationic charge at acidic urine pH in the distal nephron. This allows them to interact with anionic Tamm-Horsfall mucoprotein, thereby forming obstructing casts. However, some investigators have been unable to confirm the correlation between nephrotoxicity and pI of the light-chain proteins.
Fanconi syndrome (proximal tubular dysfunction)
Fanconi syndrome is a generalized dysfunction of the proximal tubule resulting in variable degrees of phosphate, glucose, amino acid, and bicarbonate wasting by the proximal tubule. This may occur as a hereditary disorder (in children) or as an acquired form. Acquired forms in adults are usually associated with paraproteinemias.
Light-chain proteins are catabolized in the proximal tubules, and their clearance varies inversely with the clearance of creatinine. Increased concentration of light chains exerts a toxic effect on renal tubular function; depending on the site of action, this may result in the following:
-
Fanconi syndrome (proximal tubular dysfunction)
-
Distal renal tubular acidosis
-
Nephrogenic diabetes insipidus
Light chain deposition disease
Light chain deposition disease (LCDD) is a systemic disease caused by the overproduction and extracellular deposition of monoclonal light chains. [3, 4]
Deposition does not mean pathogenicity. Deposition of light-chains similar to LCDD by IF but with no or only scanty granular electron dense deposits in the tubular basement membrane with no glomerular lesions or tubular basement membrane thickening has been described by Lin and Gallo. Hence, the IF staining of LC alone should not be considered a sufficient criteria for diagnosis of MIDD that is associated with local fibrosis.
In approximately 80% of cases, these deposits are composed of kappa, rather than lambda, light chains and are granular, and do not form fibrils or beta-pleated sheets and are negative for Congo red stain, thioflavine T, or serum amyloid protein (SAP). The constant region of the immunoglobulin light chain is deposited in this disorder, compared with the fibrils of AL amyloid that are derived from the variable region of the light chains. [5]
The pathogenesis of glomerulosclerosis in LCDD is not entirely clear, but pathogenic Ig chains stimulate mesangial cells to secrete extracellular matrix [ECM] components through growth factors, especially transforming growth factor-beta, that act as an autocoid and promote cells to produce matrix proteins, such as type IV collagen, laminin, fibronectin, and tenascin.
Myeloma kidney (cast nephropathy)
More than 50% of patients with multiple myeloma die from renal failure, and a large number of these deaths are erroneously attributed to so-called myeloma kidney. However, myeloma kidney is only one of the several causes of renal dysfunction in patients with multiple myeloma, in which specifically proteinaceous casts are observed obstructing the distal tubules and collecting ducts.
Factors that might contribute to myeloma cast nephropathy include the following:
- The direct toxicity of the intact light chains to tubular cells (compared with light chain fragment deposition in light chain deposition disease or amyloidosis)
- Protein complex formation in the distal nephron
- Tubular fluid pH
- A reduction in renal plasma flow and glomerular filtration rate (ie, decreased urine flow)
- Systemic electrolyte abnormalities (eg, hypercalcemia, hyperuricemia, hyperviscosity and dehydration)
Concomitant use of any of the following may precipitate acute kidney injury:
-
Radiocontrast agents
-
Nonsteroidal anti-inflammatory drugs
-
Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers
Amyloidosis
Adams probably recognized the association of amyloidosis and multiple myeloma in 1872, but Magnus-Levy suggested a relationship between Bence Jones proteinuria (BJP) and amyloidosis in 1931. [6]
In 1971, Glenner et al demonstrated that amyloid fibrils from a patient with primary amyloidosis had an amino acid sequence almost identical to the variable portion of monoclonal light chains (ie, Bence Jones proteins) and that amyloid fibrils could be created from Bence Jones proteins, establishing a definite link between immunoglobulin light chains and one type of amyloid. [7]
Amyloid is not a single substance, but a family of complex glycoproteins of variable composition that undergo transformation (misfolding) to from beta-pleated fibrils. Amyloids have a common characteristic ultrastructure (nonbranching fibrils 7.5-10 nm wide and of indefinite length) and tinctorial properties (green birefringence when stained with Congo red and intense yellow-green fluorescence with thioflavine T) and bind to serum amyloid P (SAP). Several forms of amyloidosis are recognized depending on nature of the precursor protein.
AL amyloid
Immunoglobulin light chains are the major constituent of these proteins, which is found in patients with primary amyloidosis and multiple myeloma. Of patients with multiple myeloma, 6-24% develop amyloidosis. Conversely, among patients presenting with primary (AL) amyloidosis, a substantial proportion have, or eventually develop, a plasma cell dyscrasia with plasmacytosis in the bone marrow, immunoglobulin light chains in the serum, and Bence-Jones proteins.
AA amyloid (SAA)
The major component of AA amyloid is a protein consisting of 76 amino acids with a molecular weight of 8500 d that is unrelated to immunoglobulins. This type is found in patients with secondary amyloidosis associated with chronic infections or inflammatory conditions, such as rheumatoid arthritis, syphilis, and chronic osteomyelitis.
Transthyretin (TTR; prealbumin)
This is wild-type or unmutated transthyretin in senile amyloidosis and a mutated form in familial amyloidosis.
Beta-2 microglobulinin dialysis-related amyloidosis
The factors that determine whether a fibrillar or granular deposition of a given monoclonal light chain are unclear and appear to be dependent on the biochemical properties or light chains and whether they are intact or in fragments. The light chains have been shown to self-associate to form high molecular weight aggregates that deposit in the tissues with or without fibril formation. Net charge of the protein may be an important determinant of amyloidogenic potential. [8]
Studies on animal models and in vitro studies of secondary (AA) amyloidosis suggest that in response to chronic injury, monocytes are activated and release interleukin 1, which acts on the liver to induce synthesis of a precursor protein designated as serum amyloid (SAA). SAA is then degraded by macrophages under the influence of certain enhancing factors, called cofactors, like serum amyloid P component (SAP), glycosaminoglycans, and certain apolipoproteins (E and J), and it is unclear if these cofactors deposit during fibrillogenesis or after a fibrillogenic event.
Etiology
Diseases frequently associated with light-chain proteinuria include mutiple myeloma, Waldenström macroglobulinemia, and amyloidosis.
Multiple myeloma (47-70%): The frequency of light-chain proteinuria depends on the type of myeloma, as follows:
-
IgG myeloma - Occurs in 60% (kappa light chain)
-
IgA myeloma - Occurs in 71% (kappa light chain)
-
IgD myeloma - Occurs in 100% (lambda light chain)
In 15% of multiple myeloma cases the clonal plasma cells are unable to produce heavy chains and instead exclusively produce light chains. Light-chain multiple myeloma has a more aggressive course and poorer prognosis than other types of multiple myeloma. [9]
Waldenström macroglobulinemia (30-40%) is usually with IgM paraproteins. IgM is a pentamer and leads to hyperviscosity syndrome. Amyloidosis (92%) is usually the lambda type.
Less frequent associations incluce malignant lymphoma, chronic lymphocytic leukemia and plasma cell leukemia. Rarer associations include nonreticular neoplasms such as angioimmunoblastic lymphadenopathy, pancreatic adenocarcinoma, and medullary thyroid carcinoma; light-chain deposition disease; and idiopathic Bence Jones proteinuria (the least common cause of light-chain proteinuria). In benign monoclonal gammopathy of unknown significance patients may have detectable light-chain proteinuria, but the amount of protein is usually negligible. Rifampin has been implicated in drug-induced light-chain proteinuria.
Light chains comprise most of the monoclonal immunoglobulin deposits in the kidney in monoclonal gammopathy of renal significance (MGRS). MGRS is defined as a B-cell or plasma cell clonal lymphoproliferation with one or more kidney lesions related to the produced monoclonal immunoglobulin, and in which the underlying clone does not cause tumor complications or meet any current hematological criteria for specific therapy. [10]
Epidemiology
The occurrence of light-chain proteinuria depends on the underlying condition. No racial predilection is recognized for this condition. Light chain–associated renal syndromes are common in men. In one study, the incidence of light-chain nephropathy was 10 times higher in men compared to women.
The incidence of monoclonal gammopathies increases with age; they occur in 1-5% of persons older than 65 years.
Overall, BJP occurs in 47-70% of persons with multiple myeloma, with the specific rate depending on the type of myeloma; with IgG myeloma, the rate is 60%; with IgA myeloma, the rate is 71%; with immunoglobulin D (IgD) myeloma, the rate is 100%. Multiple myeloma reaches a peak in the eighth decade in men, and fewer than 1% of cases are diagnosed in patients younger than 40 years.
BJB is more common in men and usually manifests in the fifth to seventh decade of life (age 40-66 y). Of patients with Waldenström macroglobulinemia, 30-40% have BJP.
Of patients with primary amyloidosis, 92% have BJP. Men are affected twice as often as women. AL amyloidosis occurs in patients older than 50 years (median age 59-63 y).
Prognosis
Overall, the prognosis depends on the type and extent of the underlying condition. Renal failure is much more prevalent in patients with light-chain proteinuria, and the severity of the renal failure correlates with the light-chain protein excretion rate. Acute renal failure is observed less frequently (8-30%), while chronic kidney disease is quite common (30-60%).
Renal insufficiency may be indolent, chronic and progressive, or rapidly progressive. Renal insufficiency, a common manifestation of multiple myeloma, is present in more than 50% of patients. [11] In one study, the risk of renal failure was 7% in patients with daily light-chain excretion of less than 0.005 g, 17%, with excretion of 0.005-2 g, and 39% with excretion of more than 2 g.
Benign monoclonal gammopathy
Clinical renal disease is uncommon in persons with true benign monoclonal gammopathy. Only 1-2% have mild renal insufficiency, and some have mild proteinuria or hematuria.
Light-chain deposition disease
The prognosis for patients with LCDD is generally poor, and death is often attributed to cardiac disease, heart failure, or infectious complications. The survival rate is 90% at 1 year and 70 % at 5 years, with renal survival in 67% and 37% at 1 and 5 years, respectively, after chemotherapy (ie, with melphalan and prednisone).
Multiple myeloma
Infections and renal failure are the major causes of death in patients with multiple myeloma. Renal failure represents the most important factor influencing survival in patients with multiple myeloma. Despite aggressive therapy, patients with renal failure and myeloma have a considerably worse prognosis compared to those with myeloma who do not have renal insufficiency. [11] The prevalence rates for renal failure are also related to the type of myeloma, ie, 14% of patients with IgG myeloma, 33% of those with IgA myeloma, and 60% of individuals with IgD myeloma have renal failure.
Factors associated with a poor prognosis in patients with multiple myeloma include high tumor mass (burden), presence of hypercalcemia (serum calcium >12 mg/dL), interstitial fibrosis and tubular atrophy based on kidney biopsy findings and pancytopenia. This is indicated by a WBC count of less than 1000/µL, a hematocrit value of less than 30%, and a platelet count less than 50,000/µL. Other indicators of poor prognosis are plasma cell leukemia, previous treatment failure(s), lambda light-chain disease compared to kappa light-chain disease, and high beta2-microglobulin level. Levels higher than 6 mcg/mL suggest a worse prognosis (survival of approximately 26 mo) compared to levels of less than 6 mcg/mL (survival of approximately 52 mo).
Once renal insufficiency is present, the relationship between the degree of renal impairment and the duration of survival is dramatic.
-
With a serum creatinine level of less than 120 µmol/L (1.4 mg/dL), median survival is 44 months.
-
With a serum creatinine level of 120-180 µmol/L (1.4-2 mg/dL), median survival is 18 months.<
-
With a serum creatinine level greater than 180 µmol/L (>2 mg/dL), median survival is 4.3 months.
Amyloidosis
The prognosis for patients with AL amyloidosis is poor, with a median survival of less than 2 years in most series. In a review of patients treated with melphalan and prednisone, the overall median survival was 89.4 months (78% 5-y survival rate) in responders versus 14.7 months (7% 5-y survival rate) in nonresponders.
Patient Education
Instruct patients to maintain adequate hydration, with a daily oral fluid intake of 2-3 liters, unless fluid restriction is needed because of advanced renal failure.
Warn patients to avoid anti-inflammatory agents because these aggravate renal dysfunction and may precipitate acute renal failure.
Educate patients about the risk of contrast agents that may precipitate kidney failure. They should question the necessity of a contrast imaging study and request alternative studies, if available.
-
Light chain–associated renal disorders. Light microscopy (hematoxylin and eosin stain at 25X power) showing nodular glomerulosclerosis (arrow) and thickening of the basement membrane. Courtesy of Madeleine Moussa, MD, FRCPC, Department of Pathology, London Health Sciences Centre, London, Ontario, Canada.
-
Light chain–associated renal disorders. Immunofluorescence (25X power) showing deposits of monotypic light chain along the basement membrane. Courtesy of Madeleine Moussa, MD, FRCPC, Department of Pathology, London Health Sciences Centre, London, Ontario, Canada.
-
Light chain–associated renal disorders. Ultrastructure (electron microscopy at 29,000X power) showing deposition of nonfibrillar electron-dense material in the mesangial nodule (arrow). Courtesy of Madeleine Moussa, MD, FRCPC, Department of Pathology, London Health Sciences Centre, London, Ontario, Canada.
-
Light chain–associated renal disorders. Ultrastructure (electron microscopy at 29,000X power) showing deposition of nonfibrillar electron-dense material along the basement membrane (arrows). Courtesy of Madeleine Moussa, MD, FRCPC, Department of Pathology, London Health Sciences Centre, London, Ontario, Canada.
-
Light chain–associated renal disorders. Immunoelectron microscopy (immunogold at 29,000X power) showing kappa light-chain deposition. Courtesy of Madeleine Moussa, MD, FRCPC, Department of Pathology, London Health Sciences Centre, London, Ontario, Canada.