Practice Essentials
Antithrombin (AT; also called heparin cofactor I; previously referred to as antithrombin III) is a 58-kDa molecule belonging to the serine protease inhibitor (serpin) superfamily that plays a central role in anticoagulation and regulating appropriate wound healing in mammalian circulation systems. Antithrombin inhibits several enzymes of the coagulation system, including factors IIa, IXa, Xa, and XIIa.
AT deficiency may be inherited or acquired.There are two types of inherited AT deficiency. Type I, which is quantitative, results from heterozygous point mutations or major gene deletions leading to low antithrombin antigen and activity levels. The more common qualitative type II AT deficiency is characterized by normal AT levels and reduced function and is further categorized into IIa, b, or c, depending on which part of the AT molecule is affected by the mutation. [1, 2] All forms of AT deficiency increase the risk for venous thrombosis and, far less commonly, arterial thrombosis.
For diagnosis, AT functional assay is the preferred initial study, to avoid missing type II deficiency. If AT function is abnormal, AT antigen levels are then measured, to distinguish between the two types of AT deficiency. Functional assays that assess inhibitory activity on Xa have a higher sensitivity than those assessing thrombin, and some patients with type II AT deficiency have only slightly reduced or even normal function, thus increasing the complexity of its diagnosis. [2]
When patients with a known inherited AT deficiency experience an acute thrombotic event and fail to respond to intravenous heparin, treatment with a direct thrombin inhibitor (eg, argatroban, dabigatran) is recommended. Correcting AT levels using antithrombin concentrate products is recommended for planned major surgery. In acute severe trauma, some studies also suggest a beneficial effect with AT replacement.
For patient education information, see Deep Vein Thrombosis.
Background
Paul Morawitz at the University of Tubingen first coined the term antithrombin in 1905 to describe plasma’s ability to neutralize thrombin activity. In 1965, Olav Egeberg described the first family with thrombotic disease due to inherited antithrombin deficiency, providing convincing evidence of the clinical importance of antithrombin. [3] For a historical overview of antithrombin research, see the excellent review by Ulrich Abildgaard. [4]
Pathophysiology
As its name implies, antithrombin was first characterized as an inhibitor of thrombin. AT also affects other serine proteases of the coagulation cascade. [5, 6, 7, 8] A diagrammatic representation of the serine proteases with which AT interacts is shown in the image below. Studies have shown that AT also has anti-inflammatory actions that are independent of its effect on regulating coagulation. [9, 10, 11, 12]
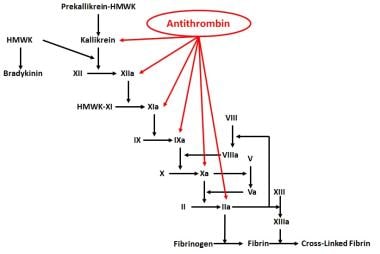
Antithrombin Function in Anticoagulation and Inflammation
AT belongs to the serpin family of inhibitors, which include heparin cofactor II (HCII), alpha2-antiplasmin, plasminogen activator inhibitor-1 (PAI-1), C1-inhibitor, and alpha1-antitrypsin. [13] AT forms a 1:1 irreversible complex with its target active enzyme, and the complex is cleared by the liver with loss of target enzyme activity.
The serpin family of proteins have a highly conserved molecular structure, with 3 beta-sheets and 9 alpha-helices. [14] A region known as the reactive center loop (RCL) protrudes above the core of the serpin molecule and has a sequence of amino acids that is complementary to binding sites in the active sites of the target proteases. Cleavage at the reactive center by target proteases results in the activation of a unique mechanism of inhibition. [15] Antithrombin exists in 2 forms: 90% as the alpha form that is glycosylated at 4 sites (Asn-96, Asn-135, Asn-155 and Asn-192) and 10% as the beta form that is not glycosylated at position Asn-135. [16]
Antithrombin is synthesized primarily in the liver. The normal plasma level is 150 mcg/mL and the plasma half-life is approximately 3 days. Thus, even short periods of abnormal liver function may reduce antithrombin production, leading to potential thrombosis.
Plasma antithrombin is comprised of 432 amino acids, 6 of which are cysteine residues that form 3 intramolecular disulfide bonds. The major physiologic role of the molecule, as the name implies, is the inhibition of thrombin (factor IIa). Additional target proteases include activated factors X, IX, XI, and XII. [17] AT also serves to reduce factor VII activity by accelerating the dissociation of the factor VIIa–tissue factor complex and preventing its reassociation. [8]
The mechanism of inactivation of serine proteinases occurs in two steps, with an initial weak interaction followed by a conformational change that ‘traps’ the protease. This mechanism is depicted in the image below.
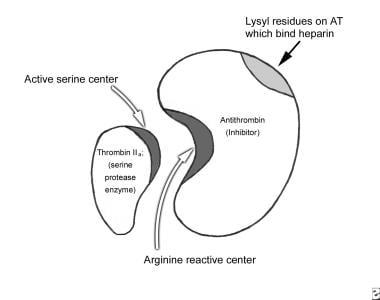 Antithrombin (AT) neutralizes the enzyme (IIa) by forming a 1:1 stoichiometric complex (AT:IIa) between the arginine-serine sites of the 2 proteins. Binding of heparin to lysyl residues on AT results in a conformational change in AT, which makes it more available to bind thrombin (IIa), IXa, and Xa, thus markedly accelerating the rate of enzyme-inhibitor complex formation. AT also neutralizes XIa and XIIa.
Antithrombin (AT) neutralizes the enzyme (IIa) by forming a 1:1 stoichiometric complex (AT:IIa) between the arginine-serine sites of the 2 proteins. Binding of heparin to lysyl residues on AT results in a conformational change in AT, which makes it more available to bind thrombin (IIa), IXa, and Xa, thus markedly accelerating the rate of enzyme-inhibitor complex formation. AT also neutralizes XIa and XIIa.
AT neutralizes factor IIa by forming a 1:1 stoichiometric complex (AT:IIa) between the arginine-serine sites of the 2 proteins. Binding of heparin to lysyl residues on AT results in a conformational change in AT, which makes it more available to bind thrombin (IIa), IXa, and Xa, thus markedly accelerating the rate of enzyme-inhibitor complex formation. AT also neutralizes factors XIa and XIIa. Transformation to the final complex involves formation of a highly stable bond between the Arg393 residue on antithrombin and the catalytic Ser residue(s) on thrombin. The formation of the antithrombin-proteinase complex is accelerated by heparin and related glycosaminoglycans, reviewed elsewhere.
In vitro studies have established the relative rates of thrombin generation and neutralization, but a study by Undas et al quantified the changes in the rate of activation and inactivation of several hemostatic factors in blood serially sampled from a bleeding time cut. [18] In this in vivo test system with an active, ongoing interaction between blood components and the injured vessel wall in flowing blood, thrombin-antithrombin (TAT) complexes started increasing within 30 seconds of the bleeding time cut and reached a maximum by 180 seconds.
The pattern of increase was typical of the two phases of activation, which have been described in other models of thrombosis, with an initial 60- to 90-second initiation phase followed by a subsequent propagation phase, during which activation reaches its maximum level. [18] In the healthy volunteers, under basal conditions, the amount of thrombin formed exceeded TAT formation at all time points tested until bleeding stopped.
TAT complexes formed following the neutralization of thrombin by antithrombin have been used as a surrogate marker for thrombin generation; serial changes in TAT levels have been used to determine alterations of the extent of hemostatic activation in the course of a disease or to assess the impact of specific therapy (eg, the effect of heparin in potentially treating disseminated intravascular coagulation).
Heparin cofactor II (HCII) is another physiologic protein inhibitor of hemostasis that appears to contribute about 20-30% of plasma heparin cofactor activity in the presence of large amounts of heparin. HCII does not, however, contribute to anti–factor Xa activity. Therefore, it has been suggested that in the assessment of the true heparin cofactor activity of antithrombin, the anti–factor Xa activity of antithrombin be measured within 30 seconds of incubation with factor Xa in the presence of small amounts of heparin in order to exclude the contribution of HCII to this assay.
The use of low doses of heparin in the test system and the use of factor Xa rather than thrombin allows for an accurate assessment of AT's heparin cofactor activity with avoidance of the contribution of HCII to this assessment. Thrombomodulin, an endothelial cell receptor for thrombin, also binds AT and accelerates its anticoagulant effect. In a purified system, tissue factor pathway inhibitor (TFPI) also appeared to potentiate the ability of AT to neutralize activated coagulation factors.
Independent of its anticoagulant properties, AT also exerts anti-inflammatory and anti-proliferative effects. A number of studies have documented the ability of AT to inhibit leukocyte rolling and adhesion, which is thought to be at least partly due to the release of prostacyclins from endothelial cells. [19]
Oelschlager et al showed that AT produces a dose-dependent reduction in both lipopolysaccharide and tumor necrosis factor (TNF)-alpha activation of nuclear factor kB (NF-kB) in cultured monocytes and endothelial cells. [20] As a result, the synthesis of proinflammatory mediators such as interleukin (IL)-6, IL-8, and TNF is decreased, leading to an anti-inflammatory effect.
A number of studies have shown that cleaved AT has potent antiangiogenic and antitumor properties. Larsson and colleagues showed that fibroblast growth factor (FGF)-induced angiogenesis in the chick embryo and angiogenesis in mouse fibrosarcoma tumors is inhibited by treatment with latent AT. [21] There is literature to suggest that latent AT may also induce apoptosis of endothelial cells by disrupting cell-matrix interactions.
Antithrombin Gene Structure
The gene for antithrombin, SERPINC1, is located on chromosome 1 band q23.1-23.9, has 7 exons and 6 introns, and is 13.5 kilobases (kb). The promoter region does not have a TATA or CAAT box. A control element at the 5' flanking region is thought to be critical for efficient synthesis of AT, with homology to an enhancer of murine and human genes. The mRNA is 1567 nucleotides long and has an approximately 175 base pair (bp) 3' untranslated region. Two modes of splicing of the primary transcript are feasible at 2 sites in the first intron; the result is either a full native AT molecule or a truncated product with a portion left within the cell.
Patients with AT deficiency, either inherited or acquired, are predisposed to venous and arterial thrombotic disease due to prolonged circulation and activity of activated coagulation factors. This increases the risk of thrombus formation at sites that fulfill Virchow's triad (stasis, alteration of coagulability of the blood, and vessel wall damage). Even a 50% reduction in the level of AT activity is sufficient to tilt the coagulation system in favor of thrombosis.
The most common thrombotic manifestations in patients with AT deficiency include lower extremity deep venous thrombosis, with recurrent VTE being common. [22] Other sites of thrombosis include the inferior vena cava, hepatic and portal veins, and renal, axillary, brachial, mesenteric, pelvic, cerebral, and retinal veins. Arterial thrombosis is far less common.
Despite the increased incidence of thrombosis, individuals with AT deficiency have a normal life expectancy. The European Prospective Cohort on Thrombophilia (EPCOT) study looked at mortality in groups of various thrombophilia patients, including AT deficiency, compared with a control group from March 1994 to December 2006. Overall, they found no increased risk of death in individuals with thrombophilia. During the study period, 6.6% of patients with AT deficiency died, compared with 5.1% of control patients (hazard ratio for death, 1.65; 95% confidence interval, 0.91–2.93). [23]
Inherited Antithrombin Deficiency
Inherited AT deficiency can be broadly classified into two types.
Type I AT deficiencies are heterozygous mutations that lead to a complete loss of the mutant antithrombin protein, resulting in immunologic and functional levels that are 50% or less than normal. The genetic basis of type I deficiencies includes major gene deletions or point mutations, with point mutations being more common. The mutations appear to cause a quantitative reduction in AT synthesis by various processes, including premature termination of translation, aberrant RNA processing, and production of unstable AT molecules that have shortened plasma half-lives. [16]
One report described 22 novel mutations in the AT gene, of which 9 missense mutations resulted in type I deficiency and led to low AT activity and antigen levels. Clinically, these mutations were all associated with venous thrombosis occurring before the age of 32 years. [24] Homozygous type I AT deficiency is almost always fatal in utero. [17]
Type II AT deficiencies are typically the result of single amino acid changes that result in functional deficits in a molecule that is otherwise normally synthesized and secreted into the plasma. The variant AT molecules may have abnormalities at the reactive site (Type IIa) or the heparin binding site (Type IIb). Most cases of type II AT deficiency are also heterozygous, although rare cases of homozygous type II deficiency have been described. [17]
A third category of type II (type IIC) AT deficiency also exists, in which multiple or "pleiotropic" abnormalities affect the reactive site, the heparin-binding site, or the plasma concentration. Type II heparin binding site variants are not associated with a high risk of thrombosis unless the affected individual is homozygous. [16]
A number of mutations in AT have been molecularly characterized. For example, the heterozygous form of a commonly inherited variant of AT affecting the heparin-binding site (HBS) is not a risk factor for thrombosis. However, several cases of patients with homozygous mutations in the HBS region of the AT gene have been published and homozygosity is associated with earlier presentation of thrombotic disease. [25] Two of these cases were shown to be associated with arterial thrombotic disease.
On the other hand, the replacement of the normal threonine-85 (Thr-85) by a nonpolar methionine (known as Antithrombin-Wibble) results in a mild adult-onset thrombotic disease, whereas replacement of the same Thr-85 by a polar lysine (known as Antithrombin-Wobble) results in early onset of thrombosis in childhood. Interestingly, fevers can trigger conformational stress on the Antithrombin-Wobble protein and favor thrombosis.
Finally, a homozygous type of AT deficiency (antithrombin III Kumamoto) has been reported in a family with consanguinity. It was shown to be associated with arterial thrombotic disease. The patient developed cerebral arterial thrombosis at age 17 years and subsequently developed venous thrombosis.
A current listing of mutations affecting the AT gene is available at the Antithrombin Mutation Database. [26] A review of published mutations indicates that they are distributed throughout the molecule, with reactive center defects having the biggest impact on the potential for thrombosis, and heparin-binding defects carrying the least thrombotic risk.
Although it is well-recognized that inherited AT deficiency confers a higher risk of coagulopathy than inherited deficiencies of protein C deficiency or protein S deficiency, there is unpredictable variability in the incidence and severity of thrombotic manifestations in patients with inherited AT deficiency. A population-based case control study found a 5-fold increased risk of thrombosis when AT deficiency was associated with another genetic defect that predisposes to thrombosis. [27, 28] This risk increased to 20-fold when AT deficiency was coupled with another acquired risk factor for thrombosis. [27] Co-inherited disorders include Activated Protein C Resistance (Factor V Leiden), protein C or S deficiency, thrombomodulin gene mutations, methylene tetrahydrofolate reductase (MTHFR) deficiency, and high lipoprotein (a) levels.
In families with inherited AT deficiency, thrombotic complications often begin in the second decade of life. Approximately 40% of these events seem to be spontaneous in nature, with no clear provoking events such as major trauma, surgery or prolonged immobility. In the remaining 60%, additional precipitating factors, such as oral contraceptive use, pregnancy, labor and delivery, surgery, or trauma, may precipitate the thrombotic event. [29]
Acquired Causes of Antithrombin Deficiency
Neonates
For healthy full-term neonates, serum AT levels are typically > 50% lower than adult reference values. Newborns do not have the thrombotic tendency noted in adults with similarly reduced values because of simultaneous reductions in their procoagulant levels and perhaps due to a protective role of alpha2-macroglobulin as a thrombin inhibitor in the neonate and in childhood. Premature infants have even lower serum levels. [30]
AT levels in the newborn rise to approximately 60% of adult levels 1 month after birth and reach normal values at approximately 3 months. [30] Genetic mutations can influence this level, but the superimposition of serious illnesses can further reduce AT due to increased consumption or decreased production.
Acute respiratory distress syndrome is a known cause of AT deficiency and itself is a major cause of both morbidity and mortality in newborns. Extracorporeal membrane oxygenation (ECMO) used in the treatment of respiratory failure can be associated with reduced AT levels and increased thrombotic events. Other causes of acquired reductions of AT in neonates include sepsis, asphyxia, liver disease, other causes of DIC, and maternal preeclampsia or eclampsia. [31, 32]
Pregnancy
There is a paucity of strong clinical evidence that reduction of AT occurs during normal pregnancy. One Scandinavian study reported that AT levels were lower during the third trimester of pregnancy and in the postpartum period, but there has been no report specifically linking thrombosis to an acquired AT deficiency. [33] Pregnancy-induced AT deficiency, however, is more likely to be seen in twin and triplet pregnancies. [34]
Diseases associated with pregnancy, such as hypertension of pregnancy, eclampsia, liver dysfunction characterized by elevations in liver enzymes, and DIC, also reduce AT levels. In these conditions, low-grade activation of coagulation with consumption of antithrombin is evident before gross deterioration of coagulation parameters occurs. [31, 35]
Pregnancy-induced AT deficiency has been shown to increase the risk of liver dysfunction independent of thrombocytopenia. [36]
Liver disease
Synthesis of AT and other physiologically important inhibitors of hemostasis, synthesis of procoagulants, and clearance of activated coagulation factors are all regulated by the liver. Thus, the liver plays a central role in hemostasis.
The severity of liver disease correlates with reductions in AT antigen levels. These reductions are not only due to impaired synthesis, but also to an element of increased consumption, particularly when additional risk factors, such as sepsis, surgery, and hypotension, are present in patients with chronic liver disease.
Patients with acute, massive hepatocellular injury and elevations of liver enzyme levels often have a significantly larger component of a consumptive process than patients with slowly progressive end-stage liver disease. Because of the decreased synthesis of inhibitors, as well as the decreased ability to clear activated coagulation factors, patients undergoing orthotopic liver transplantation predictably develop DIC with reduction in AT levels.
Kidney disease
Patients with nephrotic syndrome lose AT in the urine, resulting in reduced plasma levels and increased risk for thrombotic events. Conversely, patients with inherited AT deficiency may develop kidney failure due to renal vein thrombosis or due to glomerular deposition of fibrinogen. The degree of compromise in kidney function may be such that these patients need renal replacement therapy. Furthermore, as kidney dysfunction progresses, these patients lose increasing amounts of AT in the urine and thus become even more prone to develop thrombosis. [37, 38]
Stem cell transplantation
Veno-occlusive hepatic disease is seen in patients who undergo hematopoietic stem cell transplantation—particularly, transplantation from an unrelated donor—and it is associated with the development of microthrombi in the terminal hepatic venules. This results in rapid, marked deterioration of liver function, causing a coagulopathy characterized by the reduction in the level of AT and, consequently, significant morbidity and mortality. [39]
Sepsis
Interest in the role of AT deficiency in the setting of sepsis and the critically ill patient has been growing. There appears to be a correlation between the severity of illness and the degree of AT reduction. [17] However, the extent to which the depletion of AT affects the clinical condition of such patients, or whether a reduction in the levels of AT is merely a marker of inflammation and illness, remains to be determined.
Mesters et al in 1996 demonstrated a correlation between marked reduction in serum AT levels and poor outcomes in septic patients. [40] A number of studies thereafter suggested the use of AT supplementation in patients with severe sepsis and septic shock. [41]
However, the KyberSept trial, which was published in 2001 and was the largest randomized controlled trial of severely septic patients treated with AT supplementation, failed to demonstrate any significant beneficial effects on mortality at 28 days. [42] Of note, a subgroup of patients with severe sepsis and high risk of death with a concurrent diagnosis of DIC were found to have a significant reduction in mortality when given AT. [43]
In general, a number of studies regarding the use of AT as a treatment in the intensive care setting have overall concluded that, although there may be some benefit to such therapy, highly supraphysiologic doses of AT are necessary, and the concurrent use of any form of heparin negates the benefit that may be derived from AT treatment in this setting. [17]
More recently, Tagami et al in a large, retrospective database analysis demonstrated decreased 28-day mortality in patients with severe pneumonia and sepsis-related DIC who were given therapeutic AT. [44] Additionally, a small randomized controlled trial studying the use of AT to treat DIC in patients with sepsis demonstrated increased recovery rates from DIC, but lacked adequate power to detect a reduction in 28-day mortality. [45]
Nonetheless, the use of supplemental AT in septic patients remains controversial. Further analysis with large, randomized control studies will be required before definitive recommendations can be made.
Drug-induced reduction in antithrombin levels
Heparin, given via intravenous or subcutaneous routes, causes an approximately 30% reduction in AT levels, presumably due to rapid clearance in vivo of heparin-antithrombin complexes. Plasma samples to determine baseline AT levels must therefore not be drawn after exposure to heparin.
A large body of literature shows that estrogens/oral contraceptives can also reduce AT levels, potentially resulting in hypercoagulability (See Hypercoagulability - Hereditary Thrombophilia and Lupus Anticoagulants Associated With Venous Thrombosis and Emboli).
Finally, AT deficiency has also been described with asparaginase therapy; this occurs by suppression of production of AT in the liver as part of the mechanism of action of this chemotherapeutic agent. [46, 47]
Epidemiology
Antithrombin deficiency can be acquired and inherited. The inherited condition has a prevalence of 1 in 500–5000. [48] In patients who develop venous thrombosis, the prevalence of hereditary AT deficiency is between 1:20 and 1:200. [17] Among the subtypes of AT deficiency, type II AT deficiency is at least twice as common as type I AT deficiency in the general population. [49] However, in symptomatic patients, type I AT deficiency represents about 80% of the total cases, indicating that these individuals are more predisposed to VTE events than individuals with type II deficiencies. [50]
AT deficiency is not restricted to any particular ethnic group and has been found in many countries. In a study of 4000 Scottish blood donors, the prevalence of type I AT deficiency was found to be 0.2/1000 and that of type II heparin binding site AT deficiency was found to be 2-3/1000. [51]
Although no racial predilection for AT deficiency is known, the literature has described the presence of novel mutations in the AT gene that have been observed in thrombophilic patients in specific population groups (particularly those from the Middle East). [52, 53]
Both men and women can present with the inherited disorder and clinical manifestations of AT deficiency are evident at an early or later age, depending on the severity of the inherited genetic defect and also on the co-inheritance or presence of other thrombophilic mutations, drugs, or diseases.
The severe homozygous form of AT deficiency may lead to spontaneous fetal loss, babies born small for their gestational age due to a small placenta secondary to thrombosed placental vessels, or severe thrombotic problems at birth. In other instances, thrombotic manifestations may start in the teenage years.
Acquired AT reductions are usually secondary to other illnesses or drugs.
Prognosis
Patients who are heterozygous for type I or II AT deficiency develop significant thromboembolic complications, generally involving the deep veins. The lifetime risk of developing venous thromboembolism (VTE) depends on the subtype of AT deficiency. In patients with type I inherited AT deficiency, the risk of thrombosis is estimated to be 1% per year, starting at age 15 years. The overall lifetime risk of developing a thrombotic event in patients with type I inherited AT deficiency is estimated to range from 50% to 85%.
In patients with type II AT deficiency, the risk of developing VTE is higher in those patients who have reactive site defects as compared to heparin-binding site defects. The estimated lifetime risk of thrombosis in type II mutations has been reported to range from 6 to 20%, depending on the mutation site. [48, 16]
Patients may develop recurrent VTE at an early age and, if the condition is unrecognized or inadequately treated, they may die from such events. Long-term consequences, such as chronic leg ulcerations, severe venous varicosities, and postphlebitic syndrome, are common from repeated episodes of VTE, which cause significant morbidity. The prognosis of patients with reductions in AT as part of other systemic disorders depends on the underlying disorder.
The frequency of arterial thrombotic complications is low in patients with AT deficiency. However, mutations leading to arterial thromboses have been described.
The incidence of pregnancy-related VTE in women with AT deficiency in early reports may have overestimated due to methodologic limitations such as selection bias in family studies. Subsequent studies have suggested a much lower level with ranges between 0.08-15.8% for risk of initial VTE during. [54] However, a family history of VTE significantly increases risk during pregnancy. VTE is the leading cause of direct maternal death and thrombotic complications during embryogenesis can lead to a variety of developmental abnormalities. [2]
The risk for VTE is also increased in women taking oral contraceptives (1.2-4.4%) or hormone replacement therapy (2.5-5.1%). [54]
Nephrotic syndrome has been associated with reductions in AT and an increased incidence of venous thrombosis (renal vein, 60%; VTE, 40%) with only a 3% incidence of arterial thrombosis.
-
Antithrombin (AT) neutralizes the enzyme (IIa) by forming a 1:1 stoichiometric complex (AT:IIa) between the arginine-serine sites of the 2 proteins. Binding of heparin to lysyl residues on AT results in a conformational change in AT, which makes it more available to bind thrombin (IIa), IXa, and Xa, thus markedly accelerating the rate of enzyme-inhibitor complex formation. AT also neutralizes XIa and XIIa.
-
Cell surface–directed hemostasis (image adapted from Hoffman M, Monroe DM 3rd. A cell-based model of hemostasis. Thromb Haemost. 2001). Initially, a small amount of thrombin is generated on the surface of the tissue factor–bearing (TF-bearing) cell. Following amplification, the second burst generates a larger amount of thrombin, leading to fibrin (clot) formation.
-
Antithrombin sites of action.