Background
Wiskott-Aldrich syndrome (WAS) is an X-linked disorder characterized by the clinical triad of microthrombocytopenia, eczema, and recurrent infections. Wiskott-Aldrich syndrome is named after two physicians who originally described the condition. [1] In 1937, Alfred Wiskott, a German pediatrician, first described three brothers who had chronic bloody diarrhea, eczema, and recurrent ear infections. All three brothers died before the age of 2 years from bleeding or infection. Later, in 1954, Robert Aldrich, an American pediatrician, reported a Dutch kindred of boys who all died of similar clinical symptoms described by Wiskott, clearly demonstrating an X-linked mode of inheritance. Forty years later, the gene responsible for WAS was identified on the short arm of the X chromosome (Xp11.22-p11.23) by linkage analysis.
The gene product, Wiskott-Aldrich Syndrome Protein (WASp) is a 502 amino acid protein expressed within the cytoplasm of non-erythroid hematopoietic cells. More than 300 unique mutations in the WAS gene have been identified. The most common mutations are missense mutations, followed by nonsense, splice-site, and short deletion mutations. Of note, the original family described by Wiskott was confirmed to have a deletion of two nucleotides (AC73-74del) of the WAS gene. Depending on the mutations within the WASp gene product, there is wide variability of clinical disease. In one study of 154 patients with WAS, only 30% had the classic presentation with thrombocytopenia, small platelets, eczema, and immunodeficiency; 84% had clinical signs and symptoms of thrombocytopenia, 80% had eczema, 20% had only hematologic abnormalities, and 5% had only infectious manifestations. [2] Autoimmune disease is common and occurs in up to 40–70% of patients. There is also a significantly increased risk of lymphoreticular malignancy (10–20%), such as lymphoma, leukemia, and myelodysplasia.
In general, WAS gene mutations that cause absent protein expression result in classic WAS. Reduced WASp protein expression results in X-linked thrombocytopenia. WASp activating gain-of-function mutations result in X-linked neutropenia.
For more information, see Dermatologic Manifestations of Wiskott-Aldrich Syndrome and Pediatic Wiskott-Aldrich Syndrome.
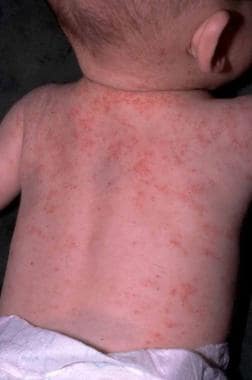 Eczematous lesions in Wiskott-Aldrich syndrome. The lesion is essentially indistinguishable from that of atopic dermatitis except for the presence of purpura and petechiae.
Eczematous lesions in Wiskott-Aldrich syndrome. The lesion is essentially indistinguishable from that of atopic dermatitis except for the presence of purpura and petechiae.
Pathophysiology
WAS results from an X-linked genetic defect in the Wiskott-Aldrich syndrome protein (WASp). The gene resides on Xp11.22-23, and its expression is limited to cells of non-erythroid hematopoietic lineage. [3] The exact function of WASp is not fully elucidated, but it seems to function as a bridge between signaling and movement of the actin filaments in the cytoskeleton. Researchers identified many different mutations [4] that interfere with the protein binding to Cdc42 and Rac GTPases, among other binding partners, most of which are involved in regulation of the actin cytoskeleton of lymphocytes. [3] This ultrastructural component of cellular architecture is involved fundamentally in intracellular and cell substrate interactions and signaling via its role in cell morphology and movement. The actin cytoskeleton is responsible for cellular functions such as growth, endocytosis, exocytosis, and cytokinesis.
Researchers propose several models of actin assembly; this topic is an extremely active area of cell biology research. [5] Actin filament growth occurs by rapid monomer addition (polymerization) to the barbed leading end of a nucleated site. Nucleation, the rate-limiting step, is stimulated by a complex of actin-related protein Arp2/3 and WASp. Cdc42 GTPase also interacts with WASp to increase this nucleation. Next, gelsolin (activated by Ca++) severs actin filaments to create barbed ends, but then must be uncapped from the filament by phosphatidylinositol 4,5-bisphosphonate and Rac to proceed with polymerization. WASp also interacts with Rac and, thus, is involved in regulation of this process at multiple interrelated sites.
WAS neutrophils have been reported to manifest abnormal NAD(P)H autofluorescence, indicating defective intracellular energy flux. Presumably, WASp mutations interfere with the proper signaling and growth of cells of the hematopoietic lineage, resulting in the platelet and immune defects observed clinically, although the exact mechanisms and defective pathways remain largely unknown.
Recently published research demonstrates that the Cdc42-WASp interaction is necessary for certain chemoattractant-induced T-cell chemotaxis. [6] Further studies have now demonstrated abnormal migration and motility in multiple key cellular components of the immune system (specifically, dendritic cells and neutrophils, as well as both B and T lymphocytes). [7, 8] With regard to WASp-deficient neutrophil adhesion and migration abnormalities, this may be caused by profound defects in clustering beta-2 integrins. [9] Also of note, CD43 (a major T-cell sialoglycoprotein) is located on microvilli; disruption of WASp regulation of cytoskeletal structure may be the cause of the CD43 defects often observed in patients with WAS. [10] WASp may also have a role in transcriptional signaling and regulation of NK cells, independent of its functions in cytoskeletal actin polymerization. [11]
Studies of genotype-phenotype correlation in WAS and closely related conditions, with detailed analyses of WASp expression, have led to our understanding of the 3 distinct WAS phenotypes: 1) classic WAS triad of microthrombocytopenia, recurrent infections, and eczema; 2) the milder X-linked thrombocytopenia variant; and 3) congenital X-linked neutropenia without any of the clinical findings chracteristic of WAS. Absent WASp protein expression results in classic WAS. Mutated WASp protein expression causes X-linked thrombocytopenia. Missense mutations at the Cdc42-binding site results in X-linked neutropenia. [12, 13, 14]
Extensive study is also underway to further identify and characterize important WASp-associated proteins, such as WASp-interacting protein (WIP) [15, 16, 17, 18, 19] and several Wiskott-Aldrich syndrome proteins verprolin homologous (WAVE). [20, 21, 22] New research suggests that N-WASp (neural Wiskott-Aldrich syndrome protein), a ubiquitously expressed homologue of WASp, may have some redundancy with WASp itself. [23]
Epidemiology
Frequency
United States
The incidence of classic WAS phenotype has been estimated at 1 and 10 in 1 million cases per live birth. Overall, WAS accounts for about 3% of all primary immunodeficiency disorders.
International
A study from Switzerland reported the incidence of WAS is 4.1 cases per 1 million live births. The same study also examined the prevalence of WAS in several national registries (ie, Italy, Japan, Switzerland, Sweden) and found that this condition occurred in 2-8.8% of patients with primary immunodeficiencies, although this statistic is subject to ascertainment bias. [24] A similar range has been documented in a national registry in Ireland, as well. [25]
Mortality/Morbidity
Median survival has increased from 8 months (patients born before 1935) to longer than 6 years (patients born after 1964). [26] In one case series, 94 surviving patients ranged in age from 1-35 years, with a median of 11 years; the average age of patients who died was 8 years. [2]
The cause of death is largely infections or bleeding, but, in one series, 12% of patients developed malignancies, primarily B-cell lymphomas, and leukemia. In that series, the relative risk of malignancy was more than 100-fold that of normal and the risk increased with age. [26] Another study showed similar results, with the reported cause of death among patients who did not receive bone marrow transplants being infection (44%), bleeding (23%), or malignancy (26%). [2]
With aggressive care (eg, splenectomy), longer survival is possible. [27, 28] Bone marrow transplant can be curative. [27] Survival rates after stem cell transplant have continued to increase, particularly after more recent emphasis on performing these procedures as soon as possible after diagnosis. [29, 30]
Outcomes of hematopoietic cell transplantation have especially improved since 2000. [31] From the most recent series of transplanted patients, overall survival from HSCT was approximately 90%.
Race-, sex-, and age-related characteristics
One large series of 301 confirmed and probable cases of WAS from 149 families reported that 8 families were black and 4 families were Chicano. Of the 40 families whose ancestry was traced outside North America, 38 emigrated from Europe. [26]
WAS is an X-linked recessive genetic disorder. The abnormal gene is relatively rare, and untreated individuals often do not survive childhood.
WAS occurs almost exclusively in males, although it is also reported in females. One report of WAS in an 8-year-old girl found a WASp gene mutation on her paternal X chromosome associated with nonrandom inactivation of her maternal X chromosome. [32] Other closely related genetic abnormalities (such as WIP deficiency) have also been reported in females, with clinical features similar to WAS. [33]
WAS is a severe congenital immunodeficiency; therefore, it occurs primarily in children. However, 2 large case series reported patients in their fourth decade of life. [26, 2]
Prognosis
Long-term prognosis was poor in the past. Prior to use of stem cell transplantation, few patients survived beyond their teens and most succumbed to compications of bleeding, infection, or malignancy. [34] Median survival in a cohort of patients born after 1964 was 6.5 years, althought survival rates have continued to increase over time. [26]
With aggressive care, prognosis has substantially improved. One study projects median survival of 25 years for patients who undergo splenectomy, and even longer for patients who undergo successful bone marrow transplant. [27] Success rates of all categories of stem cell transplantation (HLA-identical, matched/related, matched/unrelated, umbilical cord blood) have continued to climb over time. [29, 30, 31] From the most recent series of transplanted patients, overall survivial from HSCT was approximately 90%.
Patient Education
Educate patients about the function of their platelets and their immune system and about signs and symptoms that require prompt medical attention, including those seen with infections, bleeding, and malignancy.
Advise patients to appropriately restrict activities, depending on the severity of thrombocytopenia (eg, protective headgear may be indicated).
Teach patients excellent general skin care and moisturization to manage eczema.
Refer women known to be carriers for WAS for genetic counseling, and advise them that prenatal diagnosis is available.
For patient education resources, see the Skin, Hair, and Nails Center, as well as Eczema.
The Immune Deficiency Foundation (http://primaryimmune.org/) is a valuable educational resource for patients and families with primary immunodeficiency (PI) such as Wiskcott-Aldrich syndrome. There is a wealth of online educational publications and resources available to help patients and families understand and manage their condition.
The Jeffrey Modell Foundation (http://www.info4pi.org/) is another valuable online educational resource for patients and families with primary immunodeficiency. The foundation is “devoted to early and precise diagnosis, meaningful treatments, and ultimately, cures - through clinical and basic research, physician education, patient support, advocacy, public awareness and newborn screening.”
-
Eczematous lesions in Wiskott-Aldrich syndrome. The lesion is essentially indistinguishable from that of atopic dermatitis except for the presence of purpura and petechiae.




