Background
Selective immunoglobulin A deficiency (SIgAD) is a primary immunodeficiency disease and is the most common of the primary antibody deficiencies. [1] Total immunoglobulin A deficiency (IgAD) is defined as an undetectable serum immunoglobulin A (IgA) level at a value < 5 mg/dL (0.05 g/L) in humans. Partial IgAD refers to detectable but decreased IgA levels that are more than 2 standard deviations below normal age-adjusted means. [2, 3]
IgAD is commonly associated with normal B lymphocytes in peripheral blood, normal CD4+ and CD8+ T cells, and, usually, normal neutrophil and lymphocyte counts. Anti-IgA autoantibodies of the IgG and/or IgE isotype may be present. Peripheral blood may also be affected by autoimmune cytopenias, eg, autoimmune thrombocytopenia, [4, 5] and patients may have other autoimmune phenomena.
IgA was first identified by Graber and Williams in 1952; ten years later, the first patients with IgAD were described.
IgAD is a heterogeneous disorder, and the results of intensive study are beginning to elucidate genetic loci and molecular pathogenesis that contribute to various subtypes of this disorder. Several lines of evidence suggest that, in many cases, IgAD and common variable immunodeficiency (CVID) have a common pathogenesis, which is discussed further in Pathophysiology. Other data indicate different genetic risk factors. Family studies show variable inheritance patterns. Familial inheritance of IgAD occurs in approximately 20% of cases, [6] and, within families, IgAD and CVID are associated. [7, 8]
Many IgAD patients are asymptomatic (ie, "normal" blood donors) and are identified by finding a laboratory abnormality, without any apparent associated clinical disease. Some patients with IgAD may have the following associated conditions: (1) deficits in one or more immunoglobulin G (IgG) subclasses (this accounts for 20-30% of IgA-deficient patients, many of whom may have total IgG levels within the normal range) or (2) a deficient antibody response to pneumococcal immunization (specific polysaccharide antibody deficiency [SPAD]).
Some patients with IgAD later develop CVID, and family members of patients with CVID may have only selective IgAD. Characterization of the receptor for the transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI), encoded by the gene TNFRSF13B ( tumor necrosis factor receptor superfamily member 13B), suggests that people with the C104, A181E, and ins204A variants may be at risk for IgAD that progresses to CVID. [9]
Primary IgAD is permanent, and below-normal levels have been noted to remain static and persist after 20 years of observation. [10] A recent report documents a rare case of reversion. [11]
Environmental factors such as drugs or infections can cause IgAD, but this form is reversible in more than half the cases (see Causes).
Although individuals with IgAD have largely been considered healthy, recent studies indicate a higher rate of symptoms. A 20-year follow-up study that compared 204 healthy blood donors with incidentally identified IgAD to 237 healthy subjects with normal IgA levels demonstrated that 80% of IgAD donors and 50% of control subjects had episodes of infections, drug allergy, or autoimmune or atopic disease. Severe respiratory tract infections occurred in 26% of IgAD subjects, in 24% of subjects with decreased IgA levels, and in 8% of control subjects; however, the incidence of life-threatening infections was not increased. IgAD is more common in adult patients with chronic lung disease than in healthy age-matched control subjects. [12]
Patients with IgAD are at some increased risk of developing severe reactions after receiving blood products. [13, 14, 15] IgG anti-IgA antibodies may cause severe transfusion reactions if patients with IgAD are given whole blood; therefore, IgA-poor blood or washed red cells are preferred for those patients. IgA-deficient patients with immunoglobulin E (IgE)–class anti-IgA antibodies are at risk for anaphylaxis if they receive blood or intravenous immunoglobulin, but this situation is extremely rare. Individuals with such an unusual profile should receive only low IgA intravenous immunoglobulin preparations. However, caution must be used when administering IGIV to patients with IgAD if their anti-IgA status is unknown.
A history devoid of previous blood product administration does not exclude the possibility of anti-IgA antibodies or adverse reactions. Fortunately, appropriate precautions can significantly reduce morbidity (see Treatment). Blood banks can use a simple ELISA screening approach to establish an IgAD blood donor pool. [16]
Pathophysiology
IgA is the second most common immunoglobulin in human serum (after IgG) and is the predominant immunoglobulin found in mucosal secretions. Most investigators conclude that more IgA is actually produced than any other immunoglobulin, since most of it is lost in secretions.
Structurally, IgA has 2 different forms. Most serum IgA is monomeric, while secretory IgA is a dimer that contains an extra protein chain referred to as the "secretory component;" it is this property that makes this unique immunoglobulin resistant to the proteolytic enzymes found in many human secretions. The IgA dimer and the J-chain that holds the 2 IgA monomers together are produced by B cells. The secretory component is added by the serous cells in the mucoserous glands that transport dimeric IgA and other polymeric immunoglobulins (ie, IgM) onto the mucous membranes; a fragment of the receptor/transport protein mediates this translocation. Deficiency of this "polymeric Ig receptor" has been reported, and knock-out mice have been developed in the laboratory. In this situation, the serum IgA level becomes elevated because the IgA cannot be transported out of the blood into the secretions.
Secretory IgA antibodies can neutralize viruses, bind toxins, agglutinate bacteria, prevent bacteria from binding to mucosal epithelial cells, and bind to various food antigens, thus preventing their entry into the general circulation. The activities of monomeric serum IgA are not fully understood. Dimeric serum IgA probably represents a kinetically defined pool that has not yet been transported.
IgAD is a primary immunodeficiency disease presumed to result from a failure of terminal differentiation in IgA-positive B cells. The development of B-lineage cells begins in the fetal liver. B-lineage cell development then transfers to the bone marrow, when it becomes the major hematopoietic organ. Pre–B cells become immature immunoglobulin M (IgM)–positive B cells and then migrate from the bone marrow to lymph node germinal centers. After leaving the bone marrow, the B cells mature and express immunoglobulin D (IgD) receptors, respond to antigens, and, with the help of T cells (CD4+), undergo proliferation and class switching and terminal differentiation into plasma cells. [12]
In germinal centers, antigen is presented by follicular dendritic cells with help from CD4+ T cells and stimulates B cells to proliferate and undergo somatic mutation and immunoglobulin class-switching. B cells that produce high-antigen affinity antibodies are selected to develop into plasma cells that produce different immunoglobulin isotypes (ie, IgM, IgG, IgA, or IgE) or become recirculating memory B lymphocytes. These processes are regulated by cell interaction molecules (eg, CD40 on B cells, CD40 ligand on activated T cells, ICOS, TACI-TACIR, etc), and cytokines (ie, interferon-gamma and interleukin [IL]–2, IL-4, IL-5, IL-6, IL-7, IL-10, IL-12, IL-13, IL-14, and IL-15) and their cell surface receptors. [12]
Most patients with IgAD have a normal number of B cells expressing surface IgA in their blood, but the amount of surface IgA on each B cell is markedly decreased. Based on animal studies, the failure of B cells to terminally differentiate into IgA-secreting plasma cells may be due to the lack of effects caused by co-stimulatory molecules or cytokines such as IL-4, IL-6, IL-7, or IL-10.
Molecular analyses of B-cell differentiation in a small number of patients with selective or partial IgAD suggest that decreased expression level of alpha germline transcripts before a class switch might be critical for the pathogenesis of some patients with SIgAD. A case report from Japan described using reverse transcription polymerase chain reaction (RT-PCR) to separately evaluate alpha1 and alpha2 mRNAs and described a second case of alpha1 gene deletion, which manifested as a partial IgAD. [17] Another possible explanation is allelic variation in the heavy chain 3" regulatory region. A group in Canada evaluated HS1.2 allelic frequencies in 88 SIgAD patients and 101 controls, demonstrating a 39% homozygosity of the allele *1 in patients and 15% homozygosity in controls. [18]
In patients with a partial IgA deficiency, B-cell differentiation might be disturbed after a class switch. [19] The TACI receptor and 2 ligands (BAFF and APRIL) likely play a role in the pathogenesis of defective humoral immunity. BAFF is B-cell activating factor, and APRIL is a proliferation-inducing ligand. TACI is a tumor necrosis receptor superfamily member that is involved in lymphocyte maturation and survival. In mice, deletion of the sequences coding for TACI or BAFF (B-lymphocytestimulator [BLyS]) interfere with B-cell class switching and result in IgA deficiency.
Missense mutations in one allele of TACI were found in 4 of 19 unrelated individuals with CVID and in 1 of 16 individuals with SIgAD. The B cells from individuals with the TACI mutations expressed TACI but did not produce IgG and IgA in response to a TACI ligand, a finding thought to reflect impaired isotype switching. [20, 21, 6, 22, 23]
IgAD has been noted to evolve into CVID and is often observed in pedigrees that contain individuals with CVID. [24] Evidence for a common pathogenesis of CVID and IgAD include shared susceptibility alleles of major histocompatibility complex class III genes (D locus), [25] a similar spectrum of IgG subclass deficiencies, a gradual decline of immunoglobulin levels in concordant siblings, and the development of CVID in some patients with IgAD.
Previous studies of multiple-case families of patients with IgAD showed a higher prevalence of CVID among close relatives than in the general population. In multiple-case families with dominant transmission of CVID and IgAD, CVID was usually present in parents, followed by IgAD in the descendants. That study indicated the presence of a predisposing locus in the proximal part of the major histocompatibility complex. The recurrence risk was found to depend on the sex of the parents transmitting the defect. Affected mothers were more likely to produce offspring with IgAD than affected fathers. [8, 26, 27, 7]
IgAD has been reported in patients with constitutional chromosome 18 abnormalities, and a case series of 83 cases of 18p- syndrome showed an increased frequency of IgAD; however, attempts to identify a specific locus on chromosome 18 have not been successful. [7]
The ability of many patients with SIgAD to avoid respiratory infections may relate to compensatory mechanisms at the respiratory mucosal surface and/or compensatory increases in IgG. Nasal lavage samples obtained from patients with SIgAD compared to normal control show 10-fold higher median IgM levels and 3-fold higher median IgG levels. [28] An elevated nasal lavage level of the inflammatory cytokine IL-8 but not eosinophilic cationic protein (ECP) or TNF-α indicates a level of subclinical inflammation in these patients.
Nasal biopsy specimens from patients with SIgAD analyzed by Brandtzaeg et al associated a lower incidence of respiratory tract infections and an elevated ratio of IgM-producing cells to IgD-producing cells in the mucosa. [29] Patients with recurrent acute rhinosinusitis, otitis media, and tonsillitis had a dominance of the IgD over the IgM isotype, leading the investigators to conclude that immunoregulatory events favoring a dominant local IgD response did not support mucosal defense. Rose's data showed a wide range of IgM levels among patients with IgAD patients (0.87-5.2 μg/mL) but did not provide correlative clinical information. [28]
Structural lung disease such as chronic obstructive pulmonary disease (COPD) was previously thought not to associate with the ability to generate antigen-specific IgA. Studies of acute exacerbations of chronic bronchitis show that new mucosal IgA to surface-exposed epitopes of the infecting Moraxella catarrhalis isolate developed in sputum supernatants after 42% of exacerbations, [30] and significant increases in mycoplasmal-specific IgA occurred in 85% of a group of 34 patients hospitalized for acute exacerbations of COPD. In a prospective study of 250 hospitalizations for acute exacerbations of COPD, the geometric mean serum titer for IgG and IgA against Chlamydia pneumoniae was higher, with 33% meeting criteria for chronic infection. [31] In another series from India, serum and sputum IgA levels were higher in subjects with COPD than in control subjects. [32]
Recent studies, however, suggest that the mucosal IgA response is impaired in COPD with deficient transport of IgA across the bronchial epithelium, possibly involving degradation of the Ig receptor involved in transepithelial routing. [33] Like IgA deficiency, COPD/chronic bronchitis is a heterogeneous disorder, and some cases of primary defects in mucosal function (eg, cystic fibrosis) may actually be associated with increased IgA in the secretions.
Observations that SIgAD is associated with an increased prevalence of atopy suggest a possible role for IgA in asthma pathogenesis. A protective role of IgA has been seen in murine models of asthma. [33] It seems likely that in the absence of IgA, mucosal antigen exposure is increased, which may lead to increased IgE against inhalants or food antigens.
A case control study evaluated bronchial hyperresponsiveness in children with SIgAD (n = 20), children with normal IgA levels but sensitized to aeroallergens (n = 70) and children with normal IgA levels and negative skin prick tests (non-atopic) (n = 102). The children with SIgAD had lower forced vital capacity (FVC) but similar forced expiratory volume in 1 second (FEV1) values. Bronchial hyperreactivity was present in 30-35% of the children in the first 2 groups but in only 6% of the control group. The bronchial hyperreactivity among the children with SIgAD correlated with dust mite allergy but not with general atopy. [34]
Patients with partial IgAD can have diseases in which IgA is central to the pathogenesis. For example, a screening project identified 3 cases of partial IgA deficiency in patients with dermatitis herpetiformis, with IgA endomysial and tissue transglutaminase antibodies present in 2 of the patients. The authors conclude that pathogenically directed IgA antibodies were sufficient for cutaneous IgA deposition despite low serum IgA levels. [35]
Epidemiology
Frequency
United States
At a minimum, an estimated 250,000 individuals have IgAD in the United States. [36] In African Americans, the prevalence of IgAD is 1 case per 6000 persons. IgA levels are estimated to be abnormally low in 1:500 subjects, with the incidence as high as 1:100 atopic individuals. Complete absence of IgA is less frequent.
International
Factors associated with the prevalence of IgAD include a family history of IgAD and the country of origin. Family studies using IgAD blood donors as probands show that first-degree relatives have a 7.5% prevalence rate of IgAD, which is 38-fold higher than that of unrelated donors. [37] The serological prevalence of IgAD varies 100-fold among populations. Prevalences, in decreasing order, are as follows:
-
Arabian peninsula - One in 142 persons
-
Spain - One in 170 persons
-
Eastern Nigeria - One in 255 persons
-
Finland - One in 396 persons
-
Czech Republic - One in 408 persons
-
Basque regions of Spain and France - One in 521 persons
-
Canada - One in 531 persons [16]
-
Iceland - One in 533 persons
-
England - One in 875 persons
-
Brazil - One in 965 persons
-
France - One in 3040 persons
-
China (Han) - One in 2600 persons
-
China (Zhuang) - One in 5300 persons
-
Japan - One in 14,850-18,500 persons
-
Sweden - Approximately 20,000 persons affected
-
United Kingdom - Approximately 120,000 persons affected [36]
-
India - None of 3818 blood donors screened; 257 (6.7%) had partial IgAD [38]
Isolated IgAD is present in a minority of cases of transient hypogammaglobulinemia of infancy. Of a series of 40 patients presenting with recurrent responsive infections, otitis media, bronchitis or bronchial asthma, or recurrent gastroenteritis when aged 4-29 months, only 1 had isolated IgAD, 10 had reduced IgG and IgA levels, and 6 had diminished IgA and IgM levels. [39] The majority recovered immunoglobulin levels by age 3 years, but 3 had persistently low IgG and IgA levels.
A study performed by Weber-Mzell et al on 7293 healthy white volunteers demonstrated an IgAD prevalence of 0.21% (definition of IgAD was level < 0.07g/L). [40] The same study showed seasonal fluctuations of serum IgA (SIgA) concentration; levels of SIgA increased in winter.
The Latin American Group for Primary Immunodeficiency Diseases is using a registry approach to increase disorder recognition in their region. Among a total of 3321 patients registered, 53% had an antibody deficiency, of which IgAD was the more frequent phenotype. [41]
Mortality/Morbidity
IgAD is more frequent in adult subjects with chronic lung disease than in a healthy, age-matched control subjects. [12]
The 20-year longitudinal study of healthy blood donors with incidental findings of IgAD used questionnaires and medical record reviews and found a 3-fold increase in rates of severe childhood respiratory conditions (9% vs 3%), a 4-fold increase in rates of severe adult respiratory conditions (16% vs 4%), a similar increase in recurrent mild respiratory tract infections, and a significant increase in rates of recurrent viral infections (16% vs 1%).
This study also noted a 4-fold increase in the rate of autoimmune conditions (23% in subjects with SIgAD vs 5% in control subjects); a 2.5-fold increase in the rate of abdominal symptoms caused by milk (16% vs 6%); and slight increases in the rates of atopic eczema (8% vs 5%), drug allergy (9% vs 5%), and food hypersensitivity (3% vs 1%). A slight decrease was observed in the rate of allergic rhinitis and/or eczema (11% vs 17%).
When IgAD is associated with one or more IgG subclass deficiencies or impaired polysaccharide responsiveness, some individuals with IgAD may develop recurrent sinopulmonary infections, especially in patients with concurrent IgG 2 and/or 4 subclass deficiency. This condition has been found to underlie some cases of familial deafness due to severe recurrent otitis. [42] GI tract infections and disorders in patients have been reported more frequently with absent secretory IgA, as has an increased incidence of cancer. Lack of secretory IgA has been hypothesized to compromise the defense against infection with Helicobacter pylori, which is thought to be a cause of stomach cancer.
The incidence of cancer among 562 Danish and Swedish subjects with CVID or IgA was compared with that of 2017 relatives for the period 1958-1996. Among 176 subjects with CVID, the incidence of cancer (all sites) was increased (standardized incidence ratio [SIR], 1.8; 95% confidence interval [CI], 1-2.9). Stomach cancer was increased (SIR, 10.3; 95% CI, 2.1-30.2), and malignant lymphoma was increased (SIR, 12.1; 95% CI, 3.3-31). Among 386 subjects with IgAD, the incidence of cancer (all sites) was not increased (SIR, 1); however, the incidence of stomach cancer was increased, albeit to an insignificant degree (SIR, 5.4; CI, 0.7-19.5). [43] The same study did not show an increase in lymphoid malignancies (non-Hodgkin lymphoma, Hodgkin disease) in IgAD subjects, even though some evidence in the literature indicates that the risk of developing a lymphoid malignancy is increased. [44]
A recent report described 63 Israeli children with SIgAD followed for 10 years, with malignancies diagnosed in 3 children (4.8%). [45]
Patients with IgAD who have a compensatory increase in IgM in their upper respiratory tract secretions and GI fluids tend to be less symptomatic. Note that patients with total IgAD are usually more symptomatic than patients partial IgAD.
-
A previously unrecognized clear association of SIgAD with recurrent parotitis of childhood (PTC) was demonstrated by Fazekas et al in an Austrian pediatric clinic population. [46] The prevalence of PTC in IgA-deficient patients (22%) was much higher than in a large population of healthy Austrian volunteers (0.3%). [40] Case reports from other countries support this association. [47]
-
Two studies provide conflicting evidence of oral conditions in children with SIgAD. A case-controlled (n = 34 and n = 111 [control group]) study of Hungarian children showed higher decayed, missing, and filled teeth and tooth surfaces in primary dentitions. The severities of mucosal or periodontal disorders were comparable with the normal population. [48] A smaller study of Iranian children (11 cases; 11 age- and sex-matched controls) showed no differences in findings including dental caries, plaque accumulation and periodontal status. [49] A more recent case-control study of 32 Icelandic adults with SIgAD compared with 63 randomly selected controls showed similar levels of periodontal health and dental health, but higher rates of tonsillectomy (44% vs 24%), adenoidectomy (31% vs 8%), and more pharyngitis, stomatitis, and herpes labialis. [50]
Recurrent sinopulmonary infections are reported. IgAD usually manifests as recurrent otitis (in children), tonsillitis, sinusitis, and bronchitis with extracellular encapsulated bacteria (eg, Haemophilus influenzae, Streptococcus pneumoniae). Severe respiratory tract infections occur more often in adult subjects with IgAD than in normal control subjects, with a cumulative prevalence rate over 20 years of 16%. See the images below.
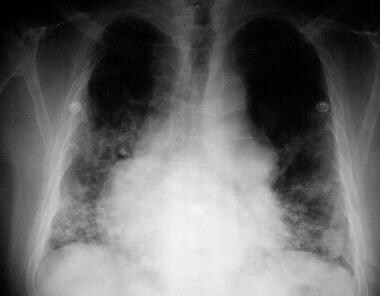 Chest radiograph of a 50-year-old man with immunoglobulin A deficiency and severe bilateral pneumonia. He also had congenital heart disease. Serum immunoglobulin G and immunoglobulin M levels were normal.
Chest radiograph of a 50-year-old man with immunoglobulin A deficiency and severe bilateral pneumonia. He also had congenital heart disease. Serum immunoglobulin G and immunoglobulin M levels were normal.
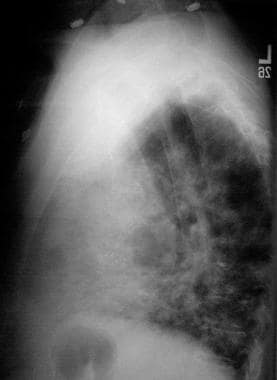 Lateral chest radiograph of a 50-year-old man with immunoglobulin A deficiency and severe bilateral pneumonia.
Lateral chest radiograph of a 50-year-old man with immunoglobulin A deficiency and severe bilateral pneumonia.
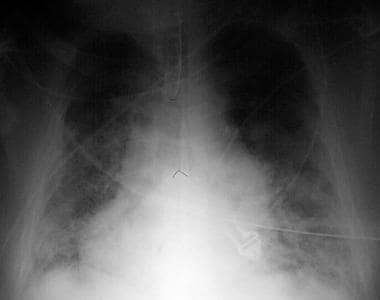 Portable chest radiograph of a 50-year-old man with acute respiratory distress syndrome as a complication of severe bilateral pneumonia. The patient died from respiratory failure 2 days after this x-ray film was taken.
Portable chest radiograph of a 50-year-old man with acute respiratory distress syndrome as a complication of severe bilateral pneumonia. The patient died from respiratory failure 2 days after this x-ray film was taken.
The substantial risk of developing lung damage, which is often reported in patients with CVID, is not a major threat to individuals who onlyhave SIgAD. In contrast, lung function is often significantly impaired among patients who have a combination of IgAD and a deficiency of one or more IgG subclasses. A few recently published cases reported the occurrence of hypersensitivity pneumonitis in patients with SIgAD and the authors suggest that SIgAD is a risk factor for a more severe course of the disease and increased susceptibility to develop extrinsic allergic alveolitis. [51, 52]
Autoimmune disease is reported in approximately 20% of patients with CVID and may be associated with IgAD.
-
Autoantibodies are often produced but may be difficult to detect. The sera of individuals with IgAD may contain various autoantibodies that cause no disease or cause myasthenia gravis or thyroid disease.
-
In Sweden, the prevalence of IgAD in thyrotropin-receptor autoantibody–seropositive individuals is 10 times higher than expected in the general population. [53]
-
Other selective case reports indicate an association between SIgAD and type 1 diabetes mellitus, vertigo, vitiligo, and alopecia.
-
Rheumatoid arthritis and systemic lupus erythematosus are the diseases most commonly connected with IgAD.
-
In a survey of serum specimens from 60 healthy subjects with SIgAD, 16 of 21 different autoantibody levels were higher in IgAD subjects than in healthy control subjects. [54]
-
A recent case report described 2 episodes of acute postinfectious glomerulonephritis, separated by 15 years, in a 33-year-old man with SIgAD. [55]
-
A northern Italian medical school described a series of 109 patients with autoimmune Addison disease (AAD), 2 of whom had SIgAD. The patients with AAD demonstrated a 12-fold higher prevalence than the local population. [56]
The prevalence rate of IgG anti-IgA antibodies among white persons with IgAD is 30-40%. In patients with combined IgA-IgG subclass 2 deficiency, the rate is 50-60%. In contrast, the prevalence of IgE antibodies against IgA is extremely low.
IgA-deficient patients with anti-IgA antibodies may develop severe reactions when they are transfused with blood components that contain IgA. In the rare cases of true anaphylaxis, these antibodies are typically of the IgE class; however, anti-IgA antibodies of the IgG isotype can also cause anaphylactic-type reactions. [57] Although anaphylactic reactions occur in 1 in 20,000-47,000 transfusions, they constitute one of the frequent nonhemolytic causes of transfusion-related mortality.
A recent case described a patient with SIgAD who developed an anaphylactic reaction to a prothrombin complex concentrate; the authors stated that product information did not list IgAD as a contraindication. [58] A recent case report described the successful management of an SIgAD patient who required massive blood transfusion during emergency cesarean delivery. [59] New data quantify the risk of allergic reactions in recipients of blood components containing anti-IgA (ie, blood donated by SIgAD donors). A Canadian donor database was cross-referenced with transfusion reaction records; the incidence of allergic reactions was 1.15% in the anti-IgA group and 2.04% in the group that did not receive anti–IgA-containing blood. [60]
GI tract infections, including chronic or recurrent giardiasis and other disorders, are reported with increased frequency. Patients with SIgAD have a 10-fold increased risk of celiac disease. Milk intolerance is common in patients with primary IgAD. Reports indicate that patients with IgAD may have IgG antibodies against cow milk and ruminant serum proteins. Patients with high titers of antibodies to cow milk reportedly are more likely to have other autoantibodies. [61] A patient with SIgAD may still mainfest IgA antibody against select targets, such as transglutaminase and endomysium, as reported in one case of celiac disease in which IgG-specific antibodies were negative. [62]
Other conditions, such as ulcerative colitis, inflammatory bowel disease, Crohn disease, and pernicious anemia, have been described in IgA-deficient individuals. Friman et al showed that individuals with SIgAD have an increased risk of becoming a carrier of E coli strains that have increased proinflammatory properties, and hypothesize that this may contribute to the development of gastrointestinal disorders in SIgAD patients. [63] Mucosal infections include acute diarrhea caused by viruses, bacteria, or Giardia lamblia parasites. A higher occurrence of serum antibodies to milk antigens in patients with IgAD suggests that normal serum IgA responses protect the host from continuing exposure to environmental antigens.
An adult sIgAD patient who worked as scaffolder and was an avocational aquarist developed concomitant cutaneous Mycobacterium haemophilum and Mycobacterium kansasii infections, infections that are typically seen only in HIV-infected or posttransplantation patients. [64]
In a case report, a girl with ring chromosome 18 (46XX, r18) had SIgAD in addition to dysmorphic features, failure to thrive, global delay of development, hypothyroidism, atopic dermatitis, bilateral chronic otitis media and aortic regurgitation with patent foramen ovale. [65]
Quality of life (QOL) studies address the cumulative impact of living with a chronic disease such as SIgAD. Several standardized scales were administered to all patients with primary antibody deficiencies seen by a single Norwegian hospital. Low QOL scores were related to unemployment, infections in more than 4 organs, and more than 2 additional diseases. Persons with SIgAD had significantly higher QOL index scores than those with other antibody deficiencies. [66]
Race
IgAD occurs in Arab persons at a rate of 1 case per 142 persons, in white persons at a rate of 1 case per 500-700 persons, in African American persons at a rate of 1 case per 6000 persons, and in Asian persons at a rate of 1 case per 14,840-18,500 persons.
Sex
A study of 7293 Austrian volunteers showed a greater frequency of SIgAD in men than in women (0.19% vs 0.014%) and a greater frequency of subnormal serum IgA levels (0.07-0.7 g/L) in men (2.66%) than in women (0.93%). [40]
Age
This disease can be diagnosed in persons of any age.
Average serum IgA levels increase 0.2 ±0.06 g/L per decade of life. [40]
-
Those older than 6 months who have recurrent upper and lower respiratory tract infections with encapsulated bacteria (eg, H influenzae, S pneumoniae) should be evaluated for IgAD as well for specific IgG antibody deficiency, although these may be maturational delays rather than fixed defects. Patients with humoral deficiencies do not usually present with recurrent infections in the first few months of life because they have circulating IgG from placental transfer.
-
Chest radiograph of a 50-year-old man with immunoglobulin A deficiency and severe bilateral pneumonia. He also had congenital heart disease. Serum immunoglobulin G and immunoglobulin M levels were normal.
-
Lateral chest radiograph of a 50-year-old man with immunoglobulin A deficiency and severe bilateral pneumonia.
-
Portable chest radiograph of a 50-year-old man with acute respiratory distress syndrome as a complication of severe bilateral pneumonia. The patient died from respiratory failure 2 days after this x-ray film was taken.




