Practice Essentials
Hyperphosphatemia—that is, abnormally high serum phosphate levels—can result from increased phosphate intake, decreased phosphate excretion, or a disorder that shifts intracellular phosphate to extracellular space. However, even severe hyperphosphatemia is for the most part clinically asymptomatic. Morbidity In patients with this condition is more commonly associated with an underlying disease than with increased phosphate values.
The image below illustrates phosphate homeostasis.
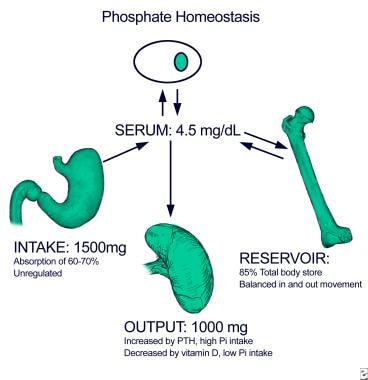 Approximately 60-70% of dietary phosphate, 1000-1500 mg/day, is absorbed in the small intestine. Although vitamin D can enhance the absorption, especially under conditions of dietary phosphate depletion, intestinal phosphate absorption does not require the presence of active vitamin D. Specifically, high serum phosphate and high dietary phosphate intake do not significantly impair intestinal uptake. The movement of phosphate in and out of bone, the reservoir containing most of the total body phosphate, is generally balanced. Renal excretion of excess dietary phosphate intake ensures maintenance of phosphate homeostasis, maintaining serum phosphate at a level of approximately 3-4 mg/dL in the serum.
Approximately 60-70% of dietary phosphate, 1000-1500 mg/day, is absorbed in the small intestine. Although vitamin D can enhance the absorption, especially under conditions of dietary phosphate depletion, intestinal phosphate absorption does not require the presence of active vitamin D. Specifically, high serum phosphate and high dietary phosphate intake do not significantly impair intestinal uptake. The movement of phosphate in and out of bone, the reservoir containing most of the total body phosphate, is generally balanced. Renal excretion of excess dietary phosphate intake ensures maintenance of phosphate homeostasis, maintaining serum phosphate at a level of approximately 3-4 mg/dL in the serum.
Signs and symptoms
Although most patients with hyperphosphatemia are asymptomatic, they occasionally report hypocalcemic symptoms, such as muscle cramps, tetany, and perioral numbness or tingling. Other symptoms include bone and joint pain, pruritus, and rash.
More commonly, patients report symptoms related to the underlying cause of the hyperphosphatemia. These generally are uremic symptoms, such as the following:
-
Fatigue
-
Shortness of breath
-
Anorexia
-
Nausea
-
Vomiting
-
Sleep disturbances
In acute hyperphosphatemia, especially that caused by parenteral phosphate administration, the patient may be hypotensive or exhibit signs of hypocalcemia, such as the following:
-
Positive Trousseau or Chvostek sign
-
Hyperreflexia
-
Carpopedal spasm
-
Seizure
See Clinical Presentation for more detail.
Diagnosis
Results from a full chemistry profile can be used as follows in determining the cause of hyperphosphatemia:
-
Low serum calcium levels along with high phosphate levels: Observed with renal failure, hypoparathyroidism, and pseudohypoparathyroidism
-
Blood urea nitrogen (BUN) and creatinine values: Help to determine whether renal failure is the cause of hyperphosphatemia
-
Elevated intact parathyroid hormone (PTH) levels: Higher likelihood in patients with renal failure or pseudohypoparathyroidism
-
Relatively low levels of intact PTH and normal renal function: Found in patients with primary or acquired hypoparathyroidism
-
High serum calcium and phosphate levels: Observed with vitamin D intoxication and milk-alkali syndrome.
-
Relatively low levels of intact PTH and high 25 and 1,25 vitamin D: Also seen in vitamin D intoxication
-
Low levels of PTH and vitamin D: Seen in milk-alkali syndrome
If renal function is normal, then more unusual disorders, such as the following, may be the cause:
-
Vitamin D intoxication
-
Laxative (Phospho-soda) abuse
-
Tumor lysis
-
Rhabdomyolysis
-
Isolated hypoparathyroidism
-
Pseudohypoparathyroidism
Rarely, if the cause of hyperphosphatemia is not clear, 24-hour measurement of urinary phosphate can be performed. Results indicate the following:
-
Fractional renal excretion exceeding 15%: Suggests either massive phosphate ingestion (eg, laxative [Phospho-soda] abuse) or lysis of tissue and resulting release of intracellular phosphate
-
Fractional renal excretion not exceeding 15%: Suggests that renal excretion is impaired because of either renal failure or hypoparathyroidism
See Workup for more detail.
Management
The major strategies for treating hyperphosphatemia are as follows:
-
Diagnose and treat the cause: Eg, hyperphosphatemia due to tumor lysis responds to forced saline diuresis to enhance urinary losses
-
Limit phosphate intake: Renal failure is the clinical condition most often requiring curtailment of phosphate ingestion; patients with advanced renal insufficiency or complete renal failure also require phosphate binders, to inhibit gastrointestinal absorption of phosphate
-
Enhance renal excretion: Used in patients with normal renal function and hyperphosphatemia; it can be accomplished most effectively by using volume repletion with saline coupled with forced diuresis with a loop diuretic such as furosemide or bumetanide
See Treatment and Medication for more detail.
Background
Phosphorus is the sixth most abundant element in the human body. A highly reactive substance, it occurs in nature, including in the human body, as phosphate.
Phosphorus (phosphate) is critical for bone mineralization, cellular structure, genetic coding, and energy metabolism. The adult body contains approximately 1000 g of phosphorus, of which 80-90% is in bone. An additional 10-14% is intracellular and the remaining 1% is extracellular.
Phosphorus is present in nearly all foods, and gastrointestinal (GI) absorption of dietary forms is very efficient. With low dietary intake, 80-90% is absorbed. When intake is greater than 10 mg/kg/day, 70% is absorbed. Normal daily dietary intake varies from 800-1500 mg.
Absorption occurs mainly in the jejunum, although some absorption occurs throughout the GI tract. A small amount of phosphorus is secreted into the GI tract. (See Etiology.)
The phosphorus in plasma is 12-17% protein bound. Free serum compounds represent much less than 1% of the total body phosphorus content. This fraction also varies with shifts between the intracellular and extracellular compartments. Thus, serum phosphorus levels may not reflect accurately the total body phosphorus content. (See Workup.)
Levels are expressed in terms of serum phosphorus mass (mg/dL). One mg/dL of phosphorus is equal to 0.32 mmol of phosphate. The normal adult range for phosphorus is 2.5-4.5 mg/dL (0.81-1.45 mmol/L). Levels are 50% higher in infants and 30% higher in children, because of growth hormone effects.
Hyperphosphatemia is considered significant when levels are greater than 5 mg/dL in adults or 7 mg/dL in children or adolescents. (See Workup and Treatment.)
Cellular function and metabolism
Phosphate is critical for a vast array of cellular processes. In addition to providing mineral strength to bone, it is an integral component of the nucleic acids that make up deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). The phosphate bonds of adenosine triphosphate (ATP) carry the energy required for all cellular functions.
The addition and deletion of phosphate groups to enzymes and proteins are common mechanisms for the regulation of their activity. Phosphate also functions as a buffer in bone, serum, and urine. In view of the sheer breadth of influence of phosphorus, phosphate homeostasis (as depicted in the image below) is understandably a highly regulated process.
Approximately 60-70% of dietary phosphate, 1000-1500 mg/day, is absorbed in the small intestine. Although vitamin D can enhance the absorption, especially under conditions of dietary phosphate depletion, intestinal phosphate absorption does not require the presence of active vitamin D. Specifically, high serum phosphate and high dietary phosphate intake do not significantly impair intestinal uptake. The movement of phosphate in and out of bone, the reservoir containing most of the total body phosphate, is generally balanced. Renal excretion of excess dietary phosphate intake ensures maintenance of phosphate homeostasis, maintaining serum phosphate at a level of approximately 3-4 mg/dL in the serum.
As previously stated, 80-90% of total body phosphate is in the bone as part of the mineralized extracellular matrix. This phosphate pool is accessible, albeit in a somewhat limited fashion. Approximately 300 mg of phosphate enters and exits bone tissue each day. Excessive losses or failure to add phosphate to bone leads to osteomalacia.
Intracellular concentration and transport
Phosphate is a predominantly intracellular anion (it has a negative charge) with a concentration of approximately 100 mmol/L, although determination of the precise intracellular concentration has been difficult. Most intracellular phosphate is either complexed or bound to proteins or lipids. In response to kinases and phosphatases, these phosphate ions attach and detach from different molecules, forming a constantly shifting pool.
Intracellular phosphate is essential for most, if not all, cellular processes; however, because the intracellular concentration of phosphate is greater than the extracellular concentration, phosphate entry into cells requires a facilitated transport process.
Several sodium-coupled transport proteins have been identified that enable intracellular uptake of phosphate by taking advantage of the steep extracellular-to-intracellular sodium gradient. Type 1 sodium phosphate cotransporters are expressed predominantly in kidney cells on the apical membranes of proximal tubule cells and liver. They are capable of transporting organic ions and stimulating chloride conductance in addition to phosphate. Their role in phosphate homeostasis is not clear. Other sites of expression include the liver and brain.
Type 2 sodium phosphate cotransporters are expressed in kidneys, bone, intestines, and a variety of other epithelial tissues responsible for transepithelial transport.
Type 2a transporters are expressed in the apical membranes of kidney proximal tubules, are very specific for phosphate, and are regulated by several physiologic mediators of phosphate homeostasis, such as parathyroid hormone (PTH), dopamine, and dietary phosphate. Currently, these transporters are believed to be most critical for maintenance of renal phosphate homeostasis. Impaired expression or function of these transporters is associated with nephrolithiasis. [1] Renal regulation of phosphate is depicted in the image below.
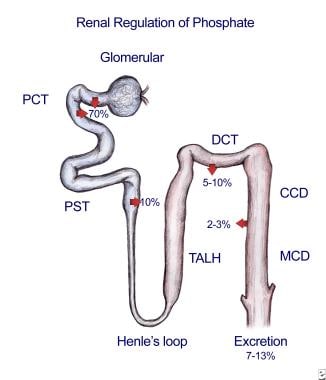 The vast majority of filtered phosphate is reabsorbed by type 2a sodium phosphate cotransporters located on the apical membrane of the renal proximal tubule. The expression of these cotransporters is increased by low dietary phosphate intake and several growth factors to enhance phosphate absorption. The expression is decreased by high dietary phosphate intake, parathyroid hormone (PTH), FGF23, and dopamine. Phosphate absorption in the remainder of the nephron is generally mediated by type 3 sodium phosphate cotransporters. No direct evidence has been found related to the regulation of these transporters in renal cells under physiologic conditions. The absorption in the proximal tubule is regulated such that the final excretion matches the dietary excess in order to maintain homeostasis.
The vast majority of filtered phosphate is reabsorbed by type 2a sodium phosphate cotransporters located on the apical membrane of the renal proximal tubule. The expression of these cotransporters is increased by low dietary phosphate intake and several growth factors to enhance phosphate absorption. The expression is decreased by high dietary phosphate intake, parathyroid hormone (PTH), FGF23, and dopamine. Phosphate absorption in the remainder of the nephron is generally mediated by type 3 sodium phosphate cotransporters. No direct evidence has been found related to the regulation of these transporters in renal cells under physiologic conditions. The absorption in the proximal tubule is regulated such that the final excretion matches the dietary excess in order to maintain homeostasis.
Type 2b transporters are very similar, but not identical, to type 2a transporters. They are expressed in the small intestine and are also upregulated under conditions of dietary phosphate deprivation. Many additional epithelial tissues express type 2b transporters, such as mammary glands, lung epithelium, salivary glands, and testis. It is unlikely that the transporters at these sites contribute to phosphate homeostasis; however, the absence of this transporter can be associated with clinical conditions such as broncholithiasis.
Type 2c transporters, a third member of the Type 2 sodium phosphate cotransporter family, were initially described as growth-related phosphate transporters. They are expressed exclusively on the S1 segment of the proximal tubule and together with Type 2a transporters are essential for normal phosphate homeostasis. Similarly to type 2a transporters, type 2c transporters are also regulated by diet and PTH. Loss of type 2c function results in hereditary hypophosphatemic rickets with hypercalciuria. [2]
Type 3 transporters were initially identified as viral transport proteins. Almost all cells express type 3 sodium phosphate cotransporters; therefore, these transporters presumably play a housekeeping role in ensuring adequate phosphate for all cells. The factors that regulate the activity of these transporter proteins are not completely understood. Evidence suggests, however, that these transporters may also participate in regulation of renal and intestinal transepithelial transport [3, 4] and in regulation of bone mineralization. [5]
Serum phosphate concentration
Circulating phosphate exists as either the univalent or divalent hydrogenated species. Because the ionization constant of acid (pK) of phosphate is 6.8, at the normal ambient serum pH of 7.4 the divalent species is 4 times as prevalent as the monovalent species.
Serum phosphate concentration varies with age, time of day, fasting state, and season. Serum phosphate concentration is higher in children than adults; the reference range is 4-7 mg/dL in children compared with 2.5-4.5 mg/dL in adults. A diurnal variation exists, with the highest phosphate level occurring near noon.
Serum phosphate concentration is regulated by diet, hormones, and physical factors such as pH. Importantly, because phosphate moves in and out of cells under several influences, the serum concentration of phosphate may not reflect true phosphate stores. Often, persons with alcoholism who have severely deficient phosphate stores may present for medical treatment with a normal serum phosphate level. Only after refeeding will serum phosphate levels decline, often abruptly plummeting to dangerously low levels.
Phosphate homeostasis
Phosphate is plentiful in the diet. A normal diet provides approximately 1000-1500 mg of phosphate, two thirds of which is absorbed, predominantly in the proximal small intestine. The fractional absorption of phosphate can be increased by increasing vitamin D intake and by ingesting a very low–phosphate diet. Under these conditions, the intestine expresses sodium-coupled phosphate transporters to enhance phosphate uptake.
Regulation of intestinal phosphate transport overall is poorly understood. Although studies had suggested that the majority of small intestine phosphate uptake was accomplished through unregulated, sodium-independent pathways, subsequent investigations have suggested that regulated sodium-dependent mechanisms may play a greater role in overall intestinal phosphate handling than was previously appreciated. Furthermore, intestinal cells may have a role in renal phosphate handling through elaboration of circulating phosphaturic substances in response to sensing a phosphate load. [6]
Absorption of phosphate can be blocked by commonly used over-the-counter aluminum-, calcium-, and magnesium-containing antacids. Mild to moderate use of such phosphate binders generally poses no threat to phosphate homeostasis, because dietary ingestion greatly exceeds body needs. However, very heavy use of these antacids can cause significant phosphate deficits. Stool losses of phosphate are minor; ie, 100-300 mg/day from sloughed intestinal cells and gastrointestinal secretions. However, diseases that cause severe diarrhea or intestinal malabsorption can dramatically increase these losses.
Bone loses approximately 300 mg of phosphate per day, but that loss is generally balanced by an uptake of 300 mg. Bone metabolism of phosphate is influenced by factors that determine bone formation and destruction; ie, PTH, vitamin D, sex hormones, acid-base balance, and inflammatory status.
PTH and vitamin D
Excess ingested phosphate is excreted by the kidneys to maintain phosphate balance. Major sites of regulation of phosphate excretion are the early proximal renal tubule and the distal convoluted tubule. In the proximal tubule, phosphate reabsorption by type 2 sodium phosphate cotransporters is regulated by dietary phosphate, PTH, and vitamin D. High dietary phosphate intake and elevated PTH levels decrease proximal renal tubule phosphate absorption, thus enhancing renal excretion. Defense against hyperphosphatemia is depicted in the image below.
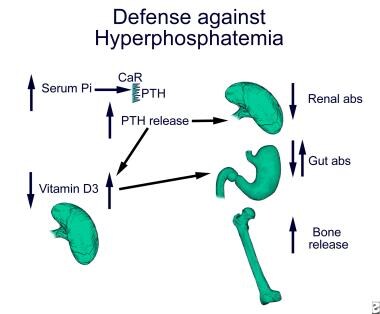 Hyperphosphatemia inhibits 1-alpha hydroxylase in the proximal tubule directly and indirectly through stimulation of FGF23, thus inhibiting the conversion of 25-hydroxy vitamin D3 to the active metabolite, 1,25 dihydroxyvitamin D3. FGF23 additionally increases the expression of 24-hydroxylase, leading to inactivation of active 1,25 dihydroxyvitamin D3. The decrease in active vitamin D production with high phosphate is somewhat offset by the ability of hyperphosphatemia to stimulate the secretion of parathyroid hormone (PTH), which will increase the activity of 1-alpha hydroxylase. The result is generally a neutral effect on intestinal phosphate absorption. Hyperphosphatemia-stimulated PTH secretion is mediated through an as yet unidentified pathway. With normal renal function, the transient increase in PTH and decrease in vitamin D serve to inhibit renal and intestinal absorption of phosphate, resulting in resolution of the hyperphosphatemia. In contrast, under conditions of renal failure, sustained hyperphosphatemia results in sustained hyperparathyroidism. The hyperparathyroidism enhances renal phosphate excretion but also enhances bone resorption, releasing more phosphate into the serum. As renal failure progresses and the ability of the kidney to excrete phosphate continues to diminish, the action of PTH on the bone can exacerbate the already present hyperphosphatemia.
Hyperphosphatemia inhibits 1-alpha hydroxylase in the proximal tubule directly and indirectly through stimulation of FGF23, thus inhibiting the conversion of 25-hydroxy vitamin D3 to the active metabolite, 1,25 dihydroxyvitamin D3. FGF23 additionally increases the expression of 24-hydroxylase, leading to inactivation of active 1,25 dihydroxyvitamin D3. The decrease in active vitamin D production with high phosphate is somewhat offset by the ability of hyperphosphatemia to stimulate the secretion of parathyroid hormone (PTH), which will increase the activity of 1-alpha hydroxylase. The result is generally a neutral effect on intestinal phosphate absorption. Hyperphosphatemia-stimulated PTH secretion is mediated through an as yet unidentified pathway. With normal renal function, the transient increase in PTH and decrease in vitamin D serve to inhibit renal and intestinal absorption of phosphate, resulting in resolution of the hyperphosphatemia. In contrast, under conditions of renal failure, sustained hyperphosphatemia results in sustained hyperparathyroidism. The hyperparathyroidism enhances renal phosphate excretion but also enhances bone resorption, releasing more phosphate into the serum. As renal failure progresses and the ability of the kidney to excrete phosphate continues to diminish, the action of PTH on the bone can exacerbate the already present hyperphosphatemia.
Conversely, low dietary phosphate intake, low PTH levels, and high vitamin D levels enhance renal proximal tubule phosphate absorption. To some extent, phosphate regulates its own regulators. High phosphate concentrations in the blood down-regulate the expression of some phosphate transporters, decrease vitamin D production, increase FGF23 release from osteocytes, and increase PTH secretion by the parathyroid gland. Distal tubule phosphate handling is less well understood. PTH increases phosphate absorption in the distal tubule, but the mechanisms by which this occurs are unknown. Renal phosphate excretion can also be increased by the administration of loop diuretics.
PTH and vitamin D were the only recognized regulators of phosphate metabolism until the discovery several novel regulators of mineral homeostasis, identified through studies of serum factors associated with phosphate-wasting syndromes, such as oncogenic osteomalacia and the hereditary forms of hypophosphatemic rickets.
PHEX
The first to be discovered was a phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX), a neutral endopeptidase mutated in the syndrome of X-linked hypophosphatemic rickets. The characteristics of this syndrome (ie, hypophosphatemia, renal phosphate wasting, low 1,25-dihydroxyvitamin D levels), as well as the fact that PHEX was identified as an endopeptidase, suggested the possibility that PHEX might be responsible for the catabolism of a non-PTH circulating factor that regulated proximal tubule phosphate transport and vitamin D metabolism. A potential substrate for PHEX was subsequently identified as fibroblast growth factor 23 (FGF23).
FGF23
Several lines of evidence support a phosphaturic role for FGF23. Another syndrome of hereditary hypophosphatemic rickets, autosomal dominant hypophosphatemic rickets, is characterized by a mutation in the FGF23 gene that renders the protein resistant to proteolytic cleavage and thus, presumably more available for inhibition of renal phosphate transport. Administration of recombinant FGF23 produces phosphaturia, and FGF23 knockout mice exhibit hyperphosphatemia.
The syndrome of oncogenic osteomalacia, characterized by acquired hypophosphatemic rickets and renal phosphate wasting in association with specific tumors, is associated with overexpression of FGF23. Interestingly, in this syndrome, overexpression of FGF23 is accompanied by 2 other phosphaturic agents; ie, matrix extracellular phosphoglycoprotein (MEPE) and frizzled related protein-4. The roles of these 2 latter proteins and their relationship with FGF23 and PHEX are unknown.
The physiologic role for FGF23 in regulation of phosphate homeostasis is still under investigation. FGF23 is produced in several tissues, including heart, liver, thyroid/parathyroid, small intestine, and bone tissue. The source of circulating FGF23 has not been conclusively established; however, the highest mRNA expression for FGF23 in mice is in bone, specifically osteocytes. [7, 8] Transgenic mouse models of FGF23 overexpression show hypophosphatemia, phosphaturia, and osteomalacia, while FGF23 deficiency is associated with hyperphosphatemia and a low fractional excretion of phosphate. FGF23 specifically decreases the proximal renal tubule expression of the type 2a and type 2c sodium phosphate cotransporters, accounting for the effect on phosphate homeostasis.
FGF23 production by osteoblasts is stimulated by 1,25 vitamin D. [8] Conversely, individuals with X-linked hypophosphatemic rickets show inappropriately depressed levels of 1,25 vitamin D due to FGF23-mediated suppression of 1-alpha hydroxylase activity. Studies in patients with end-stage renal disease found that FGF23 levels rose with decreasing creatinine clearance rates and increasing plasma phosphorus levels. As levels of 1,25 vitamin D fall during the development of progressive chronic kidney disease, levels of FGF23 rise inversely. Elevated FGF23 levels precede the development of secondary hyperparathyroidism and hyperphosphatemia in chronic kidney disease.
Klotho, a transmembrane protein, is an essential cofactor for the effects of FGF23 on renal proximal tubule cells. [9] Inactivation or deletion of Klotho expression results in hyperphosphatemia and accelerated aging. The relationship between these 2 functions of Klotho remains unknown. The loss of klotho expression occurs early in the development of chronic kidney disease. These 2 hormonal alterations, increases in FGF23 and decreases in klotho, have been associated with higher mortality and cardiovascular disease.
A study also demonstrated that FGF23 levels rapidly decreased after kidney transplantation, suggesting that FGF23 is cleared by the kidney. [10] Thus, residual FGF23 could contribute to the hypophosphatemia frequently seen in posttransplant patients. In healthy young men without renal disease, phosphate intake did not significantly increase FGF23 levels, suggesting that FGF23 may not play a role in acute phosphate homeostasis but may be more important for long-term regulation of phosphate homeostasis. [11]
STC1 and STC2
One other family of phosphate-regulating factors is the stanniocalcins (STC1 and STC2). In fish, where it was first described, STC1 inhibits calcium entry into the organism through the gills and intestines. In mammals, however, STC1 stimulates phosphate reabsorption in the small intestine and renal proximal tubules and STC2 inhibits the promoter activity of the type 2 sodium phosphate cotransporter, while the effects on calcium homeostasis are of lesser magnitude. Very little is known about the clinical significance of these newly described mineral-regulating agents or about potential interactions with either the PTH ̶ vitamin D axis or the phosphatonin-PHEX system.
Pathophysiology
Phosphorus homeostasis is normally maintained through several mechanisms. GI absorption must be matched by renal excretion, and cellular release is balanced by uptake in other tissues. Hyperphosphatemia occurs when the phosphorus load (from GI absorption, exogenous administration, or cellular release) exceeds renal excretion and tissue uptake, an imbalance that can result from any of the following three pathogenic mechanisms:
-
Excessive phosphate intake
-
Decreased phosphate excretion
-
Phosphate shift from intracellular to extracellular space
Regardless of the cause, hyperphosphatemia produces similar signs and symptoms. Because phosphate is predominantly an intracellular anion and because a variety of factors can regulate the actual serum phosphate concentration, an individual can ingest a very substantial phosphate load without exhibiting frank hyperphosphatemia. Conversely, hyperphosphatemia does not always reflect a true increase in total body phosphate stores.
Excessive phosphate intake
Excessive phosphate intake alone is an uncommon cause of hyperphosphatemia, particularly in the presence of normal renal function. The mechanisms for renal excretion allow a person with normal phosphate homeostatic mechanisms to ingest virtually unlimited quantities of phosphate. In healthy persons, higher phosphate ingestion results in higher baseline serum phosphate and higher peaks. Serum phosphate exhibits a diurnal rhythm, with the lowest concentrations being at 8 AM and the highest at 4 PM and 4 AM. Antacids decrease absorption because calcium, aluminum, and magnesium bind phosphorus into insoluble complexes. Aluminum is the most efficient binder found in antacids.
Most often, hyperphosphatemia is caused by a relatively high phosphate intake in the setting of impaired mechanisms for renal phosphate excretion (eg, renal failure, milk-alkali syndrome).
Vitamin D intoxication can produce hyperphosphatemia as a result of excessive gastrointestinal absorption and increased renal reabsorption.
Reports indicate that the excessive use of phosphate-containing laxatives or enemas can also produce hyperphosphatemia. In addition, hyperphosphatemia can result from the short-term parenteral administration of large quantities of phosphate, but again, this most often happens in the setting of impaired renal function.
Decreased phosphate excretion
Renal failure
Decreased excretion of phosphate, especially when coupled with excessive intake, is by far the most common mechanism for the development of hyperphosphatemia. The most common cause of decreased renal phosphate excretion is kidney failure, acute or chronic, of any cause (although marked hyperphosphatemia is unusual in chronic renal insufficiency unless the glomerular filtration rate (GFR) is less than 25 mL/min).
Once renal insufficiency progresses to the loss of 40-50% of renal function, the decrease in the amount of functioning renal tissue does not allow excretion of the full amount of ingested phosphate required to maintain homeostasis, and hyperphosphatemia develops.
Hyperphosphatemia may persist when patients with end-stage renal disease are placed on dialysis. Even in patients who are adherent to diet and prescribed phosphate binders, one or more of the following may be involved [12] :
-
Removal of phosphate by dialysis may vary by >400 mg per treatment
-
Enteral absorption of phosphate may differ by ≥250 mg/d among patients, even with correction for diet and vitamin D intake
-
Efficacy of phosphate binder therapy may vary 2-fold among patients
Hypoparathyroidism
Hypoparathyroidism causes hyperphosphatemia through a failure of the kidneys to inhibit renal proximal tubule phosphate reabsorption. Syndromes of tubular resistance to PTH manifest hyperphosphatemia because of the same mechanism. These syndromes include the various types of pseudohypoparathyroidism (1a, 1b, 1c, and 2) and severe hypomagnesemia, which impairs PTH secretion and causes peripheral PTH resistance.
Syndromes of tumoral calcinosis
The syndromes of tumoral calcinosis also are characterized by decreased renal excretion of phosphate, resulting in hyperphosphatemia. [13, 14, 15, 16, 17] These syndromes are produced by inactivating mutations of the following:
-
FGF23, a phosphaturic hormone
-
GALNT3, an enzyme that controls FGF23 glycosylation and function
-
Klotho, an essential cofactor for the phosphaturic effect of FGF23 in the renal tubule
Vitamin D intoxication
Vitamin D intoxication, in addition to increasing gastrointestinal phosphate absorption, increases renal phosphate reabsorption, thus enhancing the hyperphosphatemic effect.
Phosphate shift from intracellular to extracellular space
This pathogenic mechanism alone is an uncommon cause of hyperphosphatemia, but it can exacerbate hyperphosphatemia produced by impaired renal excretion. Clinical situations in which a shift to extracellular space is the major cause of hyperphosphatemia include rhabdomyolysis and tumor lysis. Rarely, extracellular shifts of phosphate occur with insulin deficiency or acute acidosis.
Sequelae of hyperphosphatemia
By precipitating calcium, decreasing vitamin D production, and interfering with PTH-mediated bone resorption, hyperphosphatemia can cause hypocalcemia; in severe cases, hypocalcemia can be life-threatening.
Prolonged hyperphosphatemia promotes soft-tissue calcification, in which an abnormal deposition of calcium phosphate occurs in previously healthy connective tissues, such as cardiac valves, and in solid organs, such as muscles.
Excess free serum phosphate is taken up into vascular smooth muscle via a type 3 sodium-phosphate cotransporter. The increased cellular phosphate activates a gene, CBFA1, that triggers a transformation in the vascular cell, causing smooth muscle cells to engage in osteogenesis. Vascular walls become calcified and arteriosclerotic, leading to increased systolic blood pressure, widened pulse pressure, and subsequent left ventricular hypertrophy.
However, although hyperphosphatemia is ultimately responsible for the increase in vascular calcifications, studies have suggested that the process may additionally be influenced by 1,25 vitamin D and an elevated calcium-phosphate product .
Related disorders
Hyperphosphatemia is an independent risk factor contributing to the increased incidence of aortic and mitral stenosis and other cardiovascular diseases among patients who are dependent on dialysis. A peripheral form known as calcific uremic arteriolopathy (calciphylaxis) can induce necrotic ulceration and gangrene in affected extremities.
Hyperphosphatemia-induced resistance to PTH contributes to secondary hyperparathyroidism and renal osteodystrophy. [18]
Cardiovascular effects
Serum phosphate level is associated with cardiovascular risk even in individuals without kidney disease in whom the serum phosphate is within the normal range. [19]
Studies have shown that acute phosphate loads obtained through dietary ingestion cause endothelial cell dysfunction, manifested as a decrease in flow-mediated dilation, in healthy men. This finding raises the possibility that the prolonged and chronic hyperphosphatemia seen in patients with chronic kidney disease could play a direct role in the enhanced cardiovascular morbidity and mortality seen in these patients. [20] A study in 70 patients who were receiving regular peritoneal dialysis found that hyperphosphatemia (as well as high-sensitivity C-reactive protein) was an independent risk factor for the initiation of coronary artery calcification. [21]
Osseocartilaginous effects
Phosphate is a major mineral component of bone; not surprisingly, therefore, chronic phosphate excess results in bone pathology, which occurs through several different mechanisms.
Some experimental evidence indicates that high phosphate levels are toxic to some cells. Specifically, a high ambient phosphate level causes apoptosis of chondrocytes and osteoblasts in cell culture. During growth, apoptosis stimulated by high phosphate levels is critical for normal bone development. [22] However, the effect of chronic hyperphosphatemia on bone and cartilage metabolism after closure of the growth plates is unknown.
Hyperphosphatemia complexes serum calcium, leading to lower-than-normal levels of ionized calcium. The decrease in ionized calcium triggers the release of PTH, resulting in a state of secondary hyperparathyroidism; high phosphate levels alone also stimulate PTH release. The elevated PTH levels lead to a high bone turnover state, resulting in the release of calcium, at the expense of bone, to normalize the serum calcium level.
High phosphate levels also inhibit 1-alpha hydroxylase, a renal enzyme that produces active vitamin D by adding a hydroxyl group to circulating 25-hydroxycholecalciferol. This inhibition is most likely a result of the hyperphosphatemia-stimulated increase in FGF23 levels.
The decrease in active vitamin D results in impaired gastrointestinal absorption of calcium, decreased renal reabsorption of calcium and phosphate, and impaired bone mineralization. Over months to years, bone density decreases. Additionally, the PTH and vitamin D derangements result in abnormal bone architecture. Clinically, the skeletal manifestations of chronic hyperphosphatemia include bone pain and fractures.
Soft-tissue calcification
Hyperphosphatemia, especially if present for an extended period, can lead to soft-tissue calcification, that is, the deposition of calcium phosphate in nonosseous sites. For example, patients with renal failure who have chronically uncontrolled hyperphosphatemia develop progressively extensive soft tissue calcifications.
Major sites of calcium deposition include the eyes, joints, and vasculature. Joint deposits can become large and painful, limiting movement and necessitating surgical removal, while eye deposits produce the syndrome of band-shaped keratopathy and conjunctivitis.
Deposition of calcium/phosphate into skin causes a papular rash and may contribute to uremic pruritus and ischemic ulcers. Calcium deposition in tendons and ligaments results in a high frequency of spontaneous rupture.
The long-term complications of chronic hyperphosphatemia can affect any organ system and are potentially devastating.
Vascular calcifications
Undoubtedly, the most significant long-term complication of chronic, uncontrolled hyperphosphatemia is the development of vascular calcifications. These can assume the following 3 basic forms:
-
Capillary and small arteriole calcifications
-
Medial arterial calcifications
-
Cardiac calcifications
Vascular calcifications produce syndromes of accelerated coronary atherosclerosis, medial arterial calcification, and calciphylaxis (which has been recognized and reported for many years in patients with renal failure).
Capillary and small arteriole deposition of calcium is generally the pathology detected in classic calciphylaxis. The blood supply distal to the calcified vessels is impaired, leading to the development of necrotic skin lesions and hemorrhagic subcutaneous lesions.
Although many case reports have been published describing the syndrome of calciphylaxis, research has been lacking; only a few series have included more than several patients. The syndrome’s pathogenesis is not known. Several investigators have suggested a role for hyperparathyroidism, excessive vitamin D, vitamin K deficiency, and high calcium phosphate production. However, many patients may not demonstrate any of these abnormalities. In contrast, most patients have a history of uncontrolled phosphate levels, implicating hyperphosphatemia as a particularly important pathogenic or inciting factor.
Medial arterial calcium deposition has been described in patients with renal failure. Some investigators suggest that smooth muscle cells in the media dedifferentiate into cells with a more osteoblastic phenotype, allowing mineralization of the blood vessel. Support for this theory comes from studies demonstrating the expression of osteoblast-specific proteins, such as alkaline phosphatase and osteopontin, in the medial cells of calcified blood vessels. Other investigators suggest that loss of normal inhibitors of soft tissue calcification, such as matrix GLA protein or osteoprotegerin, may play a role in the pathogenesis.
A study also demonstrated that phosphate uptake through Pit-1, a type III sodium-dependent phosphate cotransporter, is essential for smooth muscle cell calcification in response to elevated phosphate. Studies on coronary calcification have uniformly shown a higher degree of calcification at a younger age in patients with renal failure than in those without renal failure. This premature coronary calcification is thought to play a role in the accelerated cardiovascular mortality observed in patients with renal failure.
Calcium deposited into the heart tissue itself can disrupt the cardiac conduction system, producing significant arrhythmias. Calcium deposition into valves generally does not produce valve dysfunction, but it can serve as a marker for generalized vascular calcification. Aortic valve calcification detected using echocardiography is a poor prognostic factor in patients with renal failure and portends a high chance of mortality.
The precise role of uremia in causing, facilitating, or exacerbating the incidence and effect of vascular calcifications associated with hyperphosphatemia has not been clarified.
Etiology
The most common cause of hyperphosphatemia is renal failure. Less common causes can be classified according to pathogenesis; ie, increased phosphate intake, decreased phosphate output, or a shift of phosphate from the intracellular to the extracellular space. Often, several mechanisms contribute. Impaired renal excretion is most frequently the major factor, with relatively increased intake or cell breakdown contributing to the problem.
Increased intake
This can result from the following:
-
Excessive oral or rectal use of an oral phosphate-saline laxative (Phospho-soda)
-
Excessive parenteral administration of phosphate
-
Milk-alkali syndrome
-
Vitamin D intoxication
Medications containing phosphorus may increase intake, particularly in patients taking multiple medications. Common drugs with high phosphorus content include [23] :
-
Paroxetine
-
Amlodipine
-
Lisinopril
-
Sitagliptin
Decreased excretion
This can result from the following:
-
Acute kidney injury or chronic kidney disease
-
Hypoparathyroidism
-
Pseudohypoparathyroidism
-
Severe hypomagnesemia
-
Tumoral calcinosis
-
Bisphosphonate therapy
Shift of phosphate from intracellular to extracellular space
This can result from the following:
-
Rhabdomyolysis
-
Tumor lysis
-
Acute hemolysis
-
Acute metabolic or respiratory acidosis
Spurious
Results that falsely indicate the presence of hyperphosphatemia can result from the following:
Epidemiology
Occurrence in the United States
Hyperphosphatemia is rare in the general population, but in patients with advanced chronic kidney disease, the rate of hyperphosphatemia is at least 70%. Almost all patients with dialysis-dependent kidney failure experience hyperphosphatemia at some time during the course of their disease. This is true for acute and chronic kidney disease.
International statistics
The prevalence of hyperphosphatemia in the general population and in persons with kidney failure is similar throughout the world.
Race- and sex-related demographics
Hyperphosphatemia, per se, has no racial predilection. However, African Americans, people of Hispanic origin, and indigenous populations (eg, American Indians, aboriginal peoples) have a disproportionately high prevalence of kidney failure, which can lead to hyperphosphatemia.
Women have physiologic elevation of serum phosphate levels after menopause, but this has no known clinical significance. Notably, animal studies have shown that estrogens decrease the transcription and expression of kidney type 2a sodium phosphate cotransporters; thus, it is likely that this inhibitory effect is lost after menopause, accounting for the increase in serum phosphate. What role this mechanism may play in the shifting cardiovascular risk seen in postmenopausal women as opposed to premenopausal women has not been investigated.
Age-related demographics
Hyperphosphatemia can occur in persons of any age. The normally higher level of serum phosphate in neonates, infants, and children (sometimes >6 mg/dL) must be considered when making a diagnosis of hyperphosphatemia.
Because hyperphosphatemia most commonly occurs in the setting of kidney failure and because kidney failure occurs most commonly in elderly persons, the incidence of hyperphosphatemia increases with age, proportionate to the increase in the incidence of kidney failure. Moreover, multiple investigators have suggested that the acute and chronic kidney disease resulting from the use of phosphate-containing bowel cleansing agents is far more prevalent in the elderly population. This observation may be due to the higher prevalence of chronic kidney disease in this population.
Prognosis
Hyperphosphatemia, even of a quite severe degree, is largely a clinically asymptomatic condition. Associated morbidity most commonly results from an underlying condition than it does from the hyperphosphatemia itself.
The short-term complications of hyperphosphatemia include acute hypocalcemia with possible tetany and, more rarely, acute deposition of calcium/phosphate complexes into joints, as well as subcutaneous tissue and other areas of soft tissue. Acute hyperphosphatemia caused by excessive Phospho-soda ingestion may result in acute renal failure and, in some cases, chronic kidney disease. [29, 30, 31, 32] The inciting cause in acute hyperphosphatemia can usually be successfully treated.
In chronic hyperphosphatemia, however, the prognosis can be mixed, and the long-term complications can severely damage any organ system. The organs most commonly affected in chronic hyperphosphatemia include the vascular system, as well as the bones, skin, and heart. The joints are also commonly involved.
Prolonged hyperphosphatemia is an independent risk factor for cardiovascular disease in patients with chronic kidney disease. Patients with chronic phosphate levels above 6.5 mg/dL have an 18-39% higher mortality compared with patients with kidney failure who have near-normal serum phosphate levels.
Even in patients without chronic kidney disease, hyperphosphatemia may be a risk factor. In a Korean study of 92,756 individuals with normal kidney function, higher serum phosphorus levels were an independent predictor for all-cause mortality, particularly in men (hazard ratio 1.43, 95% confidence index, 1.22-1.68). [33]
Hyperphosphatemia in renal disease
Hyperphosphatemia is a risk factor for mortality in multiple populations, including kidney transplant recipients, [34] patients with end-stage renal disease, [35] and patients with chronic kidney disease (CKD). [36]
If started early in the course of kidney failure, control of phosphate ingestion and absorption by appropriate changes in diet and the use of binders can successfully postpone the development of various complications. This has been demonstrated most convincingly in animal studies; however, and there is a paucity of human studies. On the other hand, if hyperphosphatemia is not adequately addressed early on, the changes that occur in bones, joints, and cardiovascular tissues can be very difficult, if not impossible, to eradicate.
Changes in baseline phosphorus values beyond the recommended targets of the Kidney Disease Outcome Quality Initiative (KDOQI) have been found to be robust predictors of higher death risk. Hyperphosphatemia is associated with cardiovascular disease in CKD patients. Kidney Disease: Improving Global Outcomes (KDIGO) guidelines consider high phosphorus intake a modifiable risk factor even in the absence of hyperphosphatemia. [37]
Interestingly, whether treatment to lower phosphate levels in patients with chronic or end-stage kidney disease results in lower morbidity and mortality has not really been definitively demonstrated. A study showed that patients treated with phosphate binders had a decrease in 1-year mortality, but the effect did not correlate with the degree of hyperphosphatemia. [38] A more recent study demonstrated that when compared with placebo, treatment of patients with chronic kidney disease with one of a variety of phosphate binders resulted in lower serum phosphate but greater acceleration of vascular calcification, a condition associated with a higher mortality. [39] This is an area that requires more intensive investigation.
Patient Education
Dietary education, including on the phosphate content of foods, is very important for patients at risk for recurrent hyperphosphatemia. This information is most effectively provided by a licensed dietitian, who can provide lists of high- and low-phosphate foods and suggest substitutions when needed. [40] Patients must also be taught the importance of consistently taking phosphate binders, maintaining an adequate hydration status, and avoiding phosphorus-containing preparations, such as laxatives, enemas, and supplements.
-
Approximately 60-70% of dietary phosphate, 1000-1500 mg/day, is absorbed in the small intestine. Although vitamin D can enhance the absorption, especially under conditions of dietary phosphate depletion, intestinal phosphate absorption does not require the presence of active vitamin D. Specifically, high serum phosphate and high dietary phosphate intake do not significantly impair intestinal uptake. The movement of phosphate in and out of bone, the reservoir containing most of the total body phosphate, is generally balanced. Renal excretion of excess dietary phosphate intake ensures maintenance of phosphate homeostasis, maintaining serum phosphate at a level of approximately 3-4 mg/dL in the serum.
-
The vast majority of filtered phosphate is reabsorbed by type 2a sodium phosphate cotransporters located on the apical membrane of the renal proximal tubule. The expression of these cotransporters is increased by low dietary phosphate intake and several growth factors to enhance phosphate absorption. The expression is decreased by high dietary phosphate intake, parathyroid hormone (PTH), FGF23, and dopamine. Phosphate absorption in the remainder of the nephron is generally mediated by type 3 sodium phosphate cotransporters. No direct evidence has been found related to the regulation of these transporters in renal cells under physiologic conditions. The absorption in the proximal tubule is regulated such that the final excretion matches the dietary excess in order to maintain homeostasis.
-
Hyperphosphatemia inhibits 1-alpha hydroxylase in the proximal tubule directly and indirectly through stimulation of FGF23, thus inhibiting the conversion of 25-hydroxy vitamin D3 to the active metabolite, 1,25 dihydroxyvitamin D3. FGF23 additionally increases the expression of 24-hydroxylase, leading to inactivation of active 1,25 dihydroxyvitamin D3. The decrease in active vitamin D production with high phosphate is somewhat offset by the ability of hyperphosphatemia to stimulate the secretion of parathyroid hormone (PTH), which will increase the activity of 1-alpha hydroxylase. The result is generally a neutral effect on intestinal phosphate absorption. Hyperphosphatemia-stimulated PTH secretion is mediated through an as yet unidentified pathway. With normal renal function, the transient increase in PTH and decrease in vitamin D serve to inhibit renal and intestinal absorption of phosphate, resulting in resolution of the hyperphosphatemia. In contrast, under conditions of renal failure, sustained hyperphosphatemia results in sustained hyperparathyroidism. The hyperparathyroidism enhances renal phosphate excretion but also enhances bone resorption, releasing more phosphate into the serum. As renal failure progresses and the ability of the kidney to excrete phosphate continues to diminish, the action of PTH on the bone can exacerbate the already present hyperphosphatemia.






