Practice Essentials
Thrombotic thrombocytopenic purpura (TTP) is a rare blood disorder characterized by clotting in small blood vessels (thromboses), resulting in a low platelet count. [1, 2] In its full-blown form, the disease consists of the following pentad:
-
Microangiopathic hemolytic anemia
-
Thrombocytopenic purpura
-
Neurologic abnormalities
-
Fever
-
Kidney disease
To make an accurate diagnosis, the clinician must recognize the similarity between TTP and hemolytic-uremic syndrome (HUS). [3] In addition to HUS, the differential diagnosis includes immune thrombocytopenia (ITP) and disseminated intravascular coagulation (DIC), two entities with very different modes of therapy (see the image below).
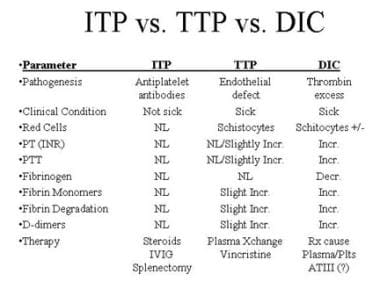 Differential diagnosis of immune thrombocytopenia (ITP), thrombotic thrombocytopenic purpura (TTP), and disseminated intravascular coagulation (DIC).
Differential diagnosis of immune thrombocytopenia (ITP), thrombotic thrombocytopenic purpura (TTP), and disseminated intravascular coagulation (DIC).
Secondary TTP has been associated with the use of certain drugs, including chemotherapy drugs such as gemcitabine and mitomycin and antiplatelet agents such as clopidogrel and ticlopidine. [4] If secondary TTP is suspected, the offending drug should be discontinued.
Signs and symptoms
TTP can affect any organ system, but involvement of the peripheral blood, the central nervous system, and the kidneys causes the clinical manifestations. Patients with TTP typically report an acute or subacute onset of symptoms related to neurologic dysfunction, anemia, or thrombocytopenia, as follows:
-
Neurologic manifestations include alteration in mental status, seizures, hemiplegia, paresthesias, visual disturbance, and aphasia
-
Fatigue may accompany the anemia
-
Severe bleeding from thrombocytopenia is unusual, although petechiae are common
See Presentation for more detail.
Diagnosis
Laboratory studies for suspected TTP include a CBC, platelet count, blood smears, coagulation studies, BUN creatinine, and serum bilirubin and lactate dehydrogenase.
The exact etiology of TTP is unknown. Most sporadic cases of TTP appear to be associated with severe deficiency of ADAMTS13 activity due to autoantibodies against this protease. [5, 6] Measuring ADAMTS13 activity level may aid in diagnosis.
Imaging studies and biopsies are not required for diagnosis.
See Workup for more detail.
Management
The therapy of choice for TTP is plasma exchange with fresh frozen plasma. Because only 20-30% of patients present with the classic pentad, initiating total plasma exchange is justified by the presence of microangiopathic hemolytic anemia (schistocytes, elevated LDH, and indirect hyperbilirubinemia) and thrombocytopenia in the absence of other obvious causes (DIC, malignant hypertension).
A recombinant ADMTS13 (Adzynma) was approved by the FDA in 2023 for prophylactic or on-demand enzyme replacement therapy in adults and children with congenital thrombotic thrombocytopenic purpura (cTTP).
Caplacizumab (Cablivi), a nanobody that targets von Willebrand factor (vWF), was approved by the FDA in 2019 for treatment of acquired thrombotic thrombocytopenic purpura (aTTP) in combination with plasma exchange and immunosuppressive therapy. It has been shown to reduce time to platelet count response and also to reduce aTTP-related death, recurrence, or major thromboembolic events. [7]
Octaplas (Octapharma), a blood plasma product extensively used in Europe, was approved by the FDA in 2013 for use in the United States. The product is a sterile, frozen solution of pooled human plasma from several donors. It is a viable alternative to single-donor plasma, and it is treated with a solvent detergent process, which reduces the risk of infection. The FDA based approval on clinical studies of patients with liver disease, liver transplant, heart surgery, and TTP. [8]
In those patients refractory to plasma exchange, using cryopoor plasma (or cryosupernatant) has sometimes led to a response. This is fresh frozen plasma that has had the cryoprecipitate removed and is thus depleted of high-molecular-weight von Willebrand multimers, which have a pathogenic role in TTP.
Corticosteroids may also be used in refractory TTP. Rituximab, although not approved for use in TTP, is increasingly recommended for use in refractory cases.
See Treatment and Medication for more detail.
Background
In 1924, Eli Moschowitz, MD, described a girl who presented with an abrupt onset of petechiae and pallor followed rapidly by paralysis, coma, and death. Upon pathologic examination, the small arterioles and capillaries of the patient were found to have thrombi consisting mostly of platelets. Dr. Moschowitz hypothesized a "powerful poison which had both agglutinative and hemolytic properties" as the cause of the disease. The syndrome described by Moschowitz is now known as thrombotic thrombocytopenic purpura (TTP). [9]
A closely related disorder, hemolytic-uremic syndrome (HUS), shares many clinical characteristics of TTP but is more common in children. Renal abnormalities tend to be more severe in HUS. Although the two disorders were once considered variants of a single syndrome, current evidence suggests differing pathogenic mechanisms of TTP and HUS. The routine use of aggressive high-volume total plasma exchange (TPE) greatly reduces the mortality of TTP, whereas the effect of TPE on the outcome of patients with HUS is more controversial.
Pathophysiology
TTP can affect any organ system, but involvement of the peripheral blood, the central nervous system, and the kidneys causes the clinical manifestations. The classic histologic lesion is one of bland thrombi in the microvasculature of affected organs. These thrombi consist predominantly of platelets, with little fibrin and red cells compared with thrombi that occur secondary to intravascular coagulation.
Patients with TTP have unusually large multimers of von Willebrand factor (vWF) in their plasma, and they have functional deficiency of a plasma protease that is responsible for the breakdown of these ultralarge vWF multimers. This protease has been isolated and cloned and is designated ADAMTS13 (A Disintegrinlike And Metalloprotease with ThromboSpondin type 1 motif 13). [10]
In more than 95% of cases, TTP is an acquired disorder that is due to autoantibodies that inhibit plasma ADAMTS13 activity; this form is termed immune-mediated TTP. [11] In the remainder of cases, TTP is an inherited disorder in which mutations in the ADAMTS13 gene result in severe deficiency of functional plasma ADAMTS13. [12] Hereditary or congenital TTP, also called Upshaw‐Schulman syndrome, accounts for less than 5% of all TTP cases but may account for 25% to 50% of cases in some patient populations, such as young children and pregnant women. [11]
In addition to immune and congenital TTP, a third form of TTP has been tentatively identified, and termed unidentified TTP. In contrast to immune TTP, anti-ADAMTS13 IgG antibodies are lacking in unidentified TPP. Significantly, whereas in patients with immune TTP, ADAMTS13 circulates in plasma in an open configuration, which makes it available for autoantibodies to bind with it, in unidentified TPP ADAMTS13 circulates in closed conformation, as is typical of healthy persons. Compared with immune TTP, unidentified TTP is less likely to occur in women, tends to occur in older individuals, and patients more often have associated cancers and less often have accompanying autoimmune diseases. [13, 14]
While ADAMTS13 deficiency seems a necessary etiologic factor, by itself it may not be sufficient to induce TTP. Endothelial activation, caused by endogenous or exogenous factors and affecting mainly microvascular cells, has been proposed as a "second hit" that triggers TTP. [15]
Although the signs and symptoms of TTP overlap with those of classic , ADAMTS13 activity is normal in most patients with classic HUS. This suggests a differing pathogenesis of these closely related entities. [16]
Epidemiology
Exact incidence figures for the United States are not available, although TTP is thought to be a rare disease. In one series, the frequency was approximately 1 in 50,000 hospital admissions. Over a 25-year period in the Sacramento, California region (population at risk, 1.2 million), at least 176 documented cases of TTP were reported. In another 1-year study, 20 institutions reported 115 patients with TTP.
Analysis of a French national registry found that the rate of TTP in France was 13 cases per million population. [6] The age-sex standardized incidence of TTP and HUS has been estimated at 2.2 cases per million population per year in the United Kingdom and 3.2 cases per million population per year in Saskatchewan, Canada. [17]
Mortality/Morbidity
Untreated, TTP has a mortality rate of as high as 90%. With plasma exchange, the mortality rate is reduced to 10-20%.
Acute morbidities include ischemic events such as stroke, transient ischemic attacks, myocardial infarction and cardiac arrhythmia, bleeding, and azotemia. TTP during pregnancy may precipitate fetal loss. [18]
In general, survivors have no long-term sequelae, with the exception of residual neurologic deficits in a minority of patients. However, relapses are not uncommon, occurring in 13-36% of patients.
Racial, Sexual, and Age-Related Disparities
An ethnic predisposition to TTP is not established. In the larger series reported, a female predominance of approximately 2:1 has been noted.
In several large studies, the median age at diagnosis is approximately 40 years. However, in the authors' series of 126 consecutive patients, the median age was 52 years.
In general, HUS is diagnosed in children and TTP is diagnosed in adults; 90% of cases of HUS occur in children. Bouw et al have presented a review article of TTP in children. [19]
-
Differential diagnosis of immune thrombocytopenia (ITP), thrombotic thrombocytopenic purpura (TTP), and disseminated intravascular coagulation (DIC).







