Practice Essentials
Disulfiram (tetraethylthiuram disulfide [TETD]) has been used for more than 50 years as a deterrent to ethanol abuse in the management of alcoholism. Approximately 200,000 alcoholics take disulfiram, or Antabuse, regularly in the United States. [1] Disulfiram has also been proposed as a deterrent to cocaine abuse, and several studies have suggested improved retention rates in treatment programs for cocaine-dependent individuals treated with disulfiram. A study found diminished "high" or "rush" after intravenous cocaine administration to healthy volunteers pretreated with disulfiram, with no change in cardiovascular parameters. [2]
The disulfiram-ethanol reaction (DER) is due to increased serum acetaldehyde concentrations generated by the metabolism of ethanol by alcohol dehydrogenase in the liver. Normally, this acetaldehyde is cleared rapidly by its metabolism to acetate via aldehyde dehydrogenase. [3] Disulfiram blocks this enzyme, irreversibly inhibiting the oxidation of acetaldehyde and causing a marked increase in acetaldehyde concentrations after ethanol consumption. The discomfort associated with this syndrome is intended to serve as a negative stimulus, but the reaction may be severe enough to cause hypotension and death.
In considering disulfiram toxicity, a distinction must be made between the clinical manifestations of a disulfiram-ethanol reaction (DER) and the toxic effects of disulfiram itself. Direct disulfiram toxicity may be further divided into acute poisoning versus chronic poisoning. The directly toxic effects of disulfiram include neurologic, cutaneous, and hepatotoxic sequelae in addition to the disulfiram-ethanol reaction.
Emergency department treatment of disulfiram-ethanol reaction (DER) is primarily supportive. See Treatment and Medication.
Pathophysiology
Ethanol is mainly metabolized in the liver to acetaldehyde by alcohol dehydrogenase (ADH). Acetaldehyde is then oxidized to acetate by aldehyde dehydrogenase (ALDH). Disulfiram irreversibly inhibits the oxidation of acetaldehyde by competing with the cofactor nicotinamide adenine dinucleotide (NAD) for binding sites on ALDH (see the image below).
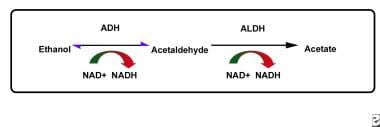 The pathway of ethanol metabolism. Disulfiram reduces the rate of oxidation of acetaldehyde by competing with the cofactor nicotinamide adenine dinucleotide (NAD) for binding sites on aldehyde dehydrogenase (ALDH).
The pathway of ethanol metabolism. Disulfiram reduces the rate of oxidation of acetaldehyde by competing with the cofactor nicotinamide adenine dinucleotide (NAD) for binding sites on aldehyde dehydrogenase (ALDH).
Ultimately, disulfiram reduces the rate of oxidation of acetaldehyde, causing a 5- to 10-fold increase in the concentration of acetaldehyde. An increased serum acetaldehyde concentration is thought to be responsible for the unpleasant side effects associated with the disulfiram-ethanol reaction.
Disulfiram also directly inhibits hepatic microsomal enzymes (cytochrome P450), in particular CYP2E1. This interferes with the metabolism of certain drugs, most notably that of warfarin, phenytoin, and theophylline. Disulfiram may also decrease the clearance of some benzodiazepines (diazepam, oxazepam, and chlordiazepoxide), caffeine, and some tricyclic antidepressants (desipramine and imipramine). The resulting possible elevation of serum concentrations of these medications has the potential to cause a corresponding toxicity.
Disulfiram is highly lipid soluble (accumulates in adipose tissue, crosses blood-brain barrier), highly protein-bound, and has 80% bioavailability after an oral dose of 350 mg. Approximately 5-20% is not metabolized and is excreted unchanged in the feces; the remainder is metabolized to both toxic and nontoxic metabolites.
The elimination of disulfiram and its numerous metabolites is a very slow process. Approximately 20% of the drug remains in the body for 1-2 weeks postingestion. Most of these metabolites are then eliminated through the gastrointestinal (GI), renal, and respiratory routes. The prolonged effects of disulfiram occur not only because the drug is slowly eliminated from the body but also because it irreversibly inhibits aldehyde dehydrogenase. In order to regain the ability to metabolize acetaldehyde, the individual must therefore synthesize new stores of the enzyme.
Disulfiram metabolites cause clinically important effects in the body (see the image below).
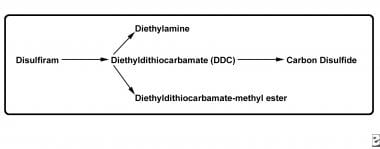
The most important toxic metabolites are diethyldithiocarbamate (DDC) and its metabolite carbon disulfide (CS2). DDC chelates copper, thus impairing the activity of dopamine beta-hydroxylase, an enzyme that catalyzes the metabolism of dopamine to norepinephrine. In this way, DDC causes depletion of presynaptic norepinephrine and accumulation of dopamine. Although hypotension from the disulfiram-ethanol reaction is mainly attributable to the effects of acetaldehyde, depletion of the potent vasoconstrictor norepinephrine may also be a contributing factor.
Dopamine agonism may be implicated in some of the altered behavior associated with disulfiram toxicity. Although no studies have directly examined the effects of low doses of disulfiram on psychotic symptoms, hypomania and psychosis have been documented in many reports among alcoholics taking high-dose disulfiram (up to 2,000 mg/d). It is possible that disulfiram, like L-dopa and amphetamine, unmasks or exacerbates preexisting psychotic symptoms in susceptible individuals by increasing central dopamine levels.
Neurotoxic effects associated with disulfiram include extrapyramidal symptoms, and lesions of the basal ganglia have been described in patients after therapy with disulfiram. Potential mechanisms for disulfiram-associated neurotoxicity include abnormal CNS metal accumulation from the chelation of copper by DDC, leading to free radical formation and neuronal oxidative stress. In addition, one study found that disulfiram and DDC increase the release of glutamate from striato-cortical synaptic vesicles, both in vitro and in rats, suggesting yet another possible mechanism for DDC-mediated neuronal damage. [4]
Other mechanisms implicated in DDC’s cytotoxic effects include its ability to chelate nickel, to interfere with sulfhydryl groups in cytochrome P-450 enzymes, and to inhibit ADH and ALDH enzymes. Furthermore, DDC inhibits superoxide dismutase, thereby impairing the ability to eliminate free radicals. DDC-induced methemoglobinemia can also occur secondary to impairment (consumption) of glutathione-dependent methemoglobin reduction.
Carbon disulfide (CS2), another disulfiram metabolite from DDC metabolism, has neurotoxic effects when administered directly. Acute exposure to CS2 causes rapid onset of headache, confusion, nausea, hallucinations, delirium, seizures, coma, and potentially death. CS2 may cause seizures by interacting with pyridoxal-5-phosphate, a cofactor in the production of GABA from glutamate, thereby depleting GABA levels in the brain and leading to benzodiazepine-resistant seizures; this forms the basis for an important experimental rat model of status epilepticus. In addition to its neurotoxic effects (neurobehavioral toxin), CS2 is hepatotoxic, inhibits cytochrome P-450, and is cardiotoxic.
The mechanism by which chronic disulfiram therapy produces hepatotoxicity is not well understood and may involve hypersensitivity or immunologic reactions in addition to the direct cytotoxic effects of its metabolites.
Etiology
Agents that may produce disulfiramlike reactions with ethanol include the following:
-
Industrial solvents ("degreasers' flush")
-
Mushrooms (eg, Coprinus atramentarius [inky cap], Clitocybe claviceps)
-
Antibiotics (eg, metronidazole, sulfonamides, some cephalosporins, nitrofurantoin, chloramphenicol)
-
Pesticides (eg, carbamates, monosulfiram [Tetmosol])
-
Chloral hydrate
-
Antifungals (griseofulvin)
Epidemiology
Disulfiram received US Food and Drug Administration (FDA) approval for use in the treatment of alcoholism in 1951. At that time, it was commonly prescribed in very high doses, up to 3,000 mg a day in some cases. This resulted in a relatively high rate of extremely severe or fatal reactions. Currently, much lower doses are used, and the incidence of disulfiram toxicity has waned. In 2020, the American Association of Poison Control Centers reported 63 single exposures, with no deaths. [5]
Prognosis
Disulfiram toxicity has a particular classification with significant overlap. The first type of toxicity is the classic disulfiram-ethanol reaction, known as the acetaldehyde syndrome. Secondly, disulfiram has its own associated acute and chronic adverse drug reactions. Finally, disulfiram-like reactions are associated with many other substances that have an ethanol-like mechanism of toxicity with disulfiram.
Disulfiram use is associated with adverse reactions at a rate of approximately 1 per 200-2000 each year. Frequently reported aversive reactions are mainly hepatic, neurologic, dermatologic, and psychiatric.
Drowsiness is the most common side effect and occurs in up to 5% of patients. It generally resolves after 2 weeks of treatment. Other side effects include dyspnea, sweating, alteration of taste, vasodilation, impotence, amblyopia, dizziness, headache, ataxia, polyneuritis, psychosis, and hypertension.
Acute disulfiram overdose is uncommon. In adults, clinical manifestations after acute overdose are rare with doses less than 3 g. Ingestion of 10-30 g may be lethal. Toxicity in children has been reported after ingestion of 2.5 g of disulfiram. Symptoms of overdose in children are mostly neurologic.
Patient Education
The patient should be advised to carry a medical alert card identifying the medication-assisted treatment, describing potential adverse effects (e.g., symptoms of a disulfiram–alcohol reaction), and providing contact information for the treating physician or institution in an emergency. [6]
Patients taking disulfiram should be advised to immediately notify their physician of any early symptoms of hepatitis, including fatigue, weakness, malaise, anorexia, nausea, vomiting, jaundice, or dark urine. [6]
For patient education resources, see Antabuse and Alcohol Use Disorder.
-
The pathway of ethanol metabolism. Disulfiram reduces the rate of oxidation of acetaldehyde by competing with the cofactor nicotinamide adenine dinucleotide (NAD) for binding sites on aldehyde dehydrogenase (ALDH).
-
Disulfiram, prodrug for active metabolites.



