Background
Stewart-Treves syndrome is a rare, deadly cutaneous angiosarcoma that develops in long-standing chronic lymphedema. [1, 2, 3, 4] See the image below.
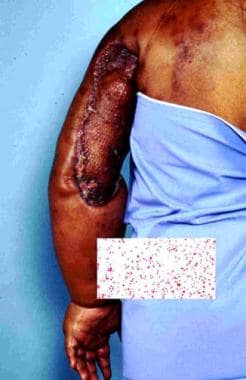 Classic example of angiosarcoma associated with chronic lymphedema after lymphadenectomy and radiation therapy.
Classic example of angiosarcoma associated with chronic lymphedema after lymphadenectomy and radiation therapy.
Most commonly, this tumor is a result of lymphedema induced by radical mastectomy to treat breast cancer. [5] Unfortunately, although the breast cancer may be cured with such radical surgery, this second primary cancer may be responsible for the patient's worsening course. The term Stewart-Treves syndrome is broadly applied to an angiosarcoma that arises in a chronically lymphedematous region due to any cause, including congenital lymphedema and other causes of secondary lymphedema unassociated with mastectomy. As reported by Durr et al in 2004, this lymphangiosarcoma occurs as a rare complication. [6] Lymphangiosarcoma is a misnomer because this malignancy seems to arise from blood vessels instead of lymphatic vessels. A more appropriate name is hemangiosarcoma.
In 1906, Lowenstein first described angiosarcoma in a patient's arm that had been affected by severe posttraumatic lymphedema for 5 years. [7] In 1948, Stewart and Treves reported this rare secondary malignancy in 6 cases of angiosarcoma in postmastectomy lymphedema. [8] They recognized that an edematous arm after radical mastectomy for breast cancer may suggest recurrent breast cancer, but that long-standing chronic edema without recurrent cancer may occasionally produce "a heretofore unrecognized and unreported sequel ... long after the malignant breast neoplasm has apparently been arrested ... a new specific tumor." Stewart and Treves suggested that these angiosarcomas were probably not observed previously because they were mistaken for recurrent, inoperable, cutaneous manifestations of breast cancer.
Lymphangiosarcoma has been described in Milroy disease and in idiopathic, congenital, traumatic, or filarial lymphedema. [9, 10, 11]
Pathophysiology
The pathogenic mechanism by which lymphedema may induce angiosarcoma has been the subject of controversy. Stewart and Treves found a high incidence of third malignancies in patients with postmastectomy angiosarcoma. Thus, they speculated that a systemic carcinogenic factor was the main causative factor in the pathogenesis of lymphangiosarcomas.
In 1979, Schreiber and others postulated the concept of local immunodeficiency in the presence of lymphedema. [12] This theory is supported by experimental evidence. In 1960, Stark and associates demonstrated that homograft skin transplanted to lymphedematous arms survive much longer than those transplanted to healthy arms. [13] Therefore, lymphedema may cause some degree of local immunodeficiency and lead to oncogenesis.
The possibility that radiation therapy has an important role in the induction of lymphangiosarcoma is also postulated. [14] Sternby et al reported that in their study, the patient with the shortest interval between radical mastectomy and the onset of the tumor (8 mo) received both preoperative radiation therapy of the breast and involved axillary lymph nodes followed by fractionated radiation. [15] Others suggest that irradiation is not an essential factor in the pathogenesis of this tumor. Finally, irradiation may be an indirect cause of lymphangiosarcomas because it may cause axillary node sclerosis and thereby accelerate and aggravate the edema.
Clinical data from Swedish women with previous breast cancer who developed angiosarcomas/lymphangiosarcomas on the thoracic wall/upper extremity between 1958 and 2008 showed 31 angiosarcomas developed at a median age of 71 years. [16] The 14 women treated for breast cancer with radical mastectomy and radiotherapy from 1949-1988 developed angiosarcomas in edematous arms after a median 11 years, whereas 17 females treated by segmental resection, antihormonal treatment, and radiotherapy from 1980-2005 developed angiosarcomas in the irradiated field on the thoracic wall after a median 7.3 years.
A hemangiogenic and lymphangiogenic origin of this angiosarcoma has been documented. [17]
Etiology
The most important single causative agent in Stewart-Treves syndrome is prolonged chronic lymphedema. Although Stewart-Treves syndrome develops after radical mastectomy in most patients, lymphangiosarcoma also develops in other forms of acquired lymphedema and in congenital lymphedema. Causes for such secondary lymphedema may include trauma, surgical invasion of the groin for the treatment of penile or cervical cancer, filariasis, idiopathic acquired lymphedema, vascular stasis, and morbid obesity.
Edema secondary to cardiac or renal disease is not associated with this malignancy. Thus, edema alone is not sufficient to cause lymphangiosarcoma. Perhaps additional factors such as a genetic predisposition are required.
Epidemiology
Frequency
Currently, approximately 400 cases of Stewart-Treves syndrome are reported in the world literature. In 1962, Schirger calculated that the incidence of this disease is 0.45% in patients who survive at least 5 years after radical mastectomy. [18] Another analysis calculated it as occurring in 0.03% of patients surviving 10 or more years after radical mastectomy. [19]
As a result of the increase in conservative treatment for breast carcinoma and improvement of operative and radiation therapy techniques, the prevalence of Stewart-Treves syndrome has decreased. [3]
Race
No racial predominance exists for Stewart-Treves syndrome.
Sex
Most patients with Stewart-Treves syndrome are women with a history of breast cancer that has been treated with radical mastectomy, which causes chronic lymphedema.
Age
Stewart-Treves syndrome usually occurs in middle-aged or elderly women, a few years or many years after mastectomy.
In 1981, Sordillo and associates reported a peak incidence in persons aged 65-70 years. [20] In 1972, Woodward et al described a series of 23 patients in a review of 163 cases of Stewart-Treves syndrome from the literature. [21] They recorded an average patient age of 68.8 years at the onset of lymphangiosarcoma; the youngest patient was aged 44 years and the oldest, 84 years.
Prognosis
The prognosis is dismal. Lymphangiosarcomas are extremely aggressive tumors with a high local recurrence rate and a tendency to metastasize early to many areas. [5]
Long-term survivors are the exceptions. The 5-year survival rate reported by Sordillo et al in 1981 was 13.6%, [20] In 1987, Hultberg found that patients with Stewart-Treves syndrome had a mean survival of 20 months after tumor onset. [22] Untreated patients have an average survival of 5-8 months. A more recent analysis showed the overall 5-year survival was 16%. [23]
Metastatic angiosarcoma to the lungs and chest wall are the most common cause of death in patients with Stewart-Treves syndrome. Metastases to the liver and bones can also occur. Lymphangiomas are associated with a high rate of local recurrence and metastasis, even after aggressive surgical treatment.
Early diagnosis and treatment by radical ablative surgery or possibly other approaches may afford an improved prognosis; patients at risk should be carefully monitored. [24, 25]
Patient Education
Patients should be informed about the significance of prolonged chronic lymphedema and about how to reduce and control it. Patients should be encouraged to seek early medical attention if they notice unexplained skin changes or unresolved lymphedema. Patients should be educated about complications, such as recurrent infections, deep venous thrombosis, and malignancies, that can occur with lymphedema.
For patient education resources, visit the Women's Health Center and Cancer Center. Also, see the patient education articles Mastectomy and Breast Cancer.
-
Classic example of angiosarcoma associated with chronic lymphedema after lymphadenectomy and radiation therapy.






