Practice Essentials
Proteus syndrome (PS) is a sporadically occurring hamartomatous disorder associated with irregular asymmetric overgrowth of multiple body tissues and cell lineages. [1] Most malformations in patients with Proteus syndrome have a mesodermal origin. Characteristic plurifocal overgrowths (partial or regional gigantism) can involve any structure of the body but most commonly involve the bone, connective tissue, and fat.
Prognosis
Patients with Proteus syndrome have difficulty ambulating because of toe macrodactyly, scoliosis, and joint instability, with frequent hip dislocations, expansive subcutaneous tumors, and compression neuropathies due to intraneural hamartomas. Some patients may have persistent atelectasis, pneumonia, or symptoms of pulmonary insufficiency. Intellectual disability is present in approximately 30% of patients. Premature death has been reported in 20% of Proteus syndrome patients, most often related to deep venous thrombosis leading to pulmonary embolus, postoperative complications, or pneumonia. [2, 3]
The disease is progressive but somewhat variable in prognosis. With appropriate medical and surgical care, patients with Proteus syndrome may age normally. [4] However, despite treatment efforts, Proteus syndrome may result in extreme musculoskeletal, cutaneous, and visceral deformities. [5]
Signs and symptoms
See Physical Examination.
Patients present with the characteristic abnormalities of Proteus syndrome, many of which are not present at birth. By definition, these findings are progressive and asymmetric. This is a sporadic disorder, and the patients would not have a family history of similar disorders.
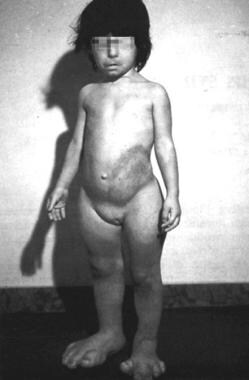
Complications
Sequelae in patients with Proteus syndrome include ambulatory difficulty due to toe macrodactyly, scoliosis, and joint instability, with frequent hip dislocations.
Dysregulated adipose growth can result in aggressive, infiltrating lipomas that can involve adjacent structures.
Pulmonary complications are a frequent cause of morbidity and mortality in Proteus syndrome patients.
Diagnostics
Imaging studies are helpful to establish the diagnosis of Proteus syndrome and in tracking the progression of the disease. The following imaging studies may be useful:
-
Radiography of the skull, vertebral column, long bones, and pelvis: The most characteristic forms of overgrowth are macrodactyly, clinodactyly, asymmetrical hypertrophy of a limb, vertebral body abnormalities, and hyperostosis. [6]
-
Comparative radiographic study of the hands and feet: The most characteristic non-cutaneous features are progressive asymmetric macrodactyly, hemihypertrophy of any part of the skeleton, scoliosis and spinal canal stenosis, macrocephaly, and exostoses, especially of the skull.
-
Intracranial MRI: This is an essential initial examination to evaluate for malformations of the CNS that may be associated with intellectual disability or seizures. Such findings may include multiple meningiomas, polymicrogyria, and periventricular heterotopias.
-
Abdominal MRI: Even in the absence of symptoms, abdominal MRI is important to exclude intra-abdominal lipomas. If present, these can behave aggressively, invading adjacent structures.
-
High-resolution chest CT scanning: This examination may be useful in evaluating pulmonary cystic malformations.
Skin biopsy: Skin biopsy should be considered to confirm the clinical diagnosis of a cerebriform connective-tissue nevus. See Proteus syndrome diagnostic criteria, category A, in Physical Examination. The histologic findings in Proteus syndrome are specific to the particular type of lesion. The histology of cerebriform connective-tissue nevi and epidermal nevi are discussed in Physical Examination, along with a description of their clinical manifestations.
Genetic testing is available for the AKT1 gene.
Electroencephalography: Although most patients appear to be of normal intelligence, intellectual disability may occur. Seizures have also been reported.
The ongoing evaluation of the patient with Proteus syndrome should include the following procedures [7] :
-
Serial clinical photography
-
Consultation with a dermatologist, followed by biopsy if indicated
-
Consultation with an orthopedic surgeon, followed by surgical treatment if indicated
-
Ongoing treatment from a geneticist, pediatrician, or both
-
Consultations with a neurologist and an ophthalmologist
-
Referral to family support group
Management
The aim of medical treatment is to minimize the physical and psychosocial consequences of Proteus syndrome; this requires a multidisciplinary approach. [8]
Antithrombotic prophylaxis should be considered if the patient is undergoing a surgical procedure because the patient's vascular malformations predispose them to deep venous thromboses and fatal pulmonary emboli. [9]
Cystic pulmonary lesions should be followed closely because they may progress to pneumonia, atelectasis and potentially pulmonary insufficiency. Pulmonary hamartomas can impair respiratory function. One report describes the successful reduction in hamartoma size with rapamycin (sirolimus) therapy. [10]
Cerebriform connective-tissue nevi are of considerable concern for patients, especially when present on the plantar surfaces, making walking uncomfortable. Conservative medical treatment for cerebriform connective-tissue nevi includes (1) keeping the feet clean and dry, (2) regularly applying antibacterial lotion to reduce odor, (3) closely monitoring for ulceration and infection, and (4) using orthotic devices. Attempts at surgical excision have led to poor outcomes; therefore, surgery is not recommended. [11]
Sirolimus has been used for the management of Proteus syndrome in a child with encouraging results. [12] Miransertib (MK-7075), a pan-AKT inhibitor, is under trial for the management of Proteus syndrome. [13]
Also see Surgical Care and Consultations.
Background
Although some evidence of this syndrome was published in the medical literature as early as 1907, the modern medical description of the disease is attributed to Cohen and Hayden, who identified the syndrome in 1979. [14] In 1983, to stress the polymorphic nature of the clinical manifestations of the disorder, Wiedermann named it "Proteus syndrome" after the Greek god Proteus, who could change his shape at will to avoid capture. [15]
Perhaps the most famous case of Proteus syndrome is that of Joseph Merrick, the "Elephant Man" described by Sir Frederick Treves in 1884, who was made famous by a stage play and movie of the same name. Although first thought to have neurofibromatosis, Merrick is now believed to have had Proteus syndrome. Preserved castings of his soles show cerebriform cutaneous hyperplasia, a characteristic finding in persons with Proteus syndrome. [16]
Pathophysiology
Proteus syndrome is a rare, sporadic disease with patchy or mosaic manifestations. [17] In 2011, the New England Journal of Medicine published a paper by Lindhurst et al naming a mutation in AKT1 as the cause of Proteus syndrome. [18] Twenty-nine patients with Proteus syndrome were studied, and 26 were found to have activation of AKT protein in the affected tissues. All unaffected patients were negative for the activating mutation.
Patients with Proteus syndrome have a somatic mutation, meaning that the mutation arises randomly during development of the fetus after fertilization. Only tissues that descend from the mutated cell have the AKT activation and, therefore, have phenotypic abnormalities. Patients who have mutation at an earlier stage of development will then have more severe clinical symptoms.
The mutated AKT1 is constitutively activated as a result of phosphorylation. AKT is part of the PI3K/AKT/MTOR pathway, which regulates apoptosis of tissues. Activation of AKT leads to lack of apoptosis and overgrowth of tissues.
While PTEN was previously thought to be implicated in this disorder, this is no longer thought to be the case. Patients with PTEN mutation are now thought to have Proteus-like syndrome. [19, 20]
Etiology
Proteus syndrome is a rare, sporadic disease with patchy or mosaic manifestations. In 2011, the New England Journal of Medicine published a paper by Lindhurst et al naming a mutation in AKT1 as the cause of Proteus syndrome. [18, 21] Twenty-nine patients with Proteus syndrome were studied, and 26 were found to have activation of AKT protein in the affected tissues. All unaffected patients were negative for the activating mutation.
Patients with Proteus syndrome have a somatic mutation, meaning that the mutation arises randomly during development of the fetus after fertilization. Only tissues that descend from the mutated cell have the AKT activation and, therefore, have phenotypic abnormalities. Patients who have mutation at an earlier stage of development will then have more severe clinical symptoms.
The mutated AKT1 is constitutively activated as a result of phosphorylation. AKT is part of the PI3K/AKT/MTOR pathway, which regulates apoptosis of tissues. Activation of AKT leads to lack of apoptosis and overgrowth of tissues.
While PTEN was previously thought to be implicated in this disorder, this is no longer thought to be the case. Patients with PTEN mutation are now thought to have Proteus-like syndrome. [19, 20]
Epidemiology
Around 100 cases have been reported in the literature. [22] Proteus syndrome has no predilection for any particular race. The male-to-female ratio is 1.9:1 (n = 96). [2]
Age
At least some of the abnormalities associated with Proteus syndrome can be present at birth or appear in the first years of life. Frank asymmetry or overgrowth is present in less than 18%. Symptoms usually progress until puberty, when there seems to be a plateau. [23] Notably, the cerebriform nevi typically does not manifest until age 2 years, on average, often delaying the correct diagnosis of Proteus syndrome. [24]
-
Epidermal nevus of the trunk follows the Blaschko lines.
-
Cerebriform connective-tissue nevus on the plantar surface.
-
Overall clinical aspect.
-
Proteus syndrome with hemihypertrophy of the limbs.
-
Proteus syndrome with gigantism of the feet and macrodactyly.









