Practice Essentials
Lipoid proteinosis is a rare autosomal recessive genodermatosis characterized by the deposition of an amorphous hyaline material in the skin, mucosa, and viscera. It is also known as Urbach-Wiethe disease or cutaneous-mucosal hyalinosis. Consanguinity in patients' parents is identified in approximately 20% of lipid proteinosis cases. [1] The classic manifestation is onset in infancy with a hoarse cry due to laryngeal infiltration. Skin and mucous membrane changes become apparent clinically, and the disease typically follows a slowly progressive, yet often benign, course. Virtually any organ may be involved, but visceral involvement rarely leads to clinically significant consequences. The exceptions are involvement of the central nervous system and respiratory tract, which may result in seizures and airway obstruction, respectively. Lifespan is otherwise normal.
Lipoid proteinosis has been linked to mutations in the gene encoding extracellular matrix protein 1 (ECM1). [2, 3, 4] To date, no effective treatment is known.
See the image below:
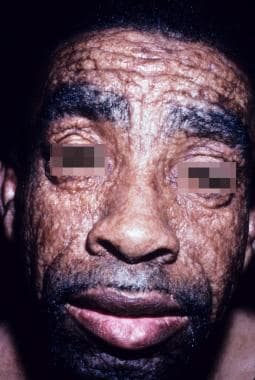 Adult male with lipoid proteinosis. His leonine facies appearance is a result of diffuse skin infiltration. Courtesy of Kenneth E. Greer, MD.
Adult male with lipoid proteinosis. His leonine facies appearance is a result of diffuse skin infiltration. Courtesy of Kenneth E. Greer, MD.
Prognosis
Lipoid proteinosis has a stable or slowly progressive course, and early manifestations are predictive of outcome. [5]
The presence of this disease is compatible with a normal life span unless altered by airway obstruction or fatal seizure activity.
Mortality rates in infants and adults are slightly increased because of laryngeal obstruction. Patients with significant airway compromise may require permanent tracheostomy.
Causes
Loss of function mutations in the gene encoding extracellular matrix protein 1 (ECM1) on band 1q21 has been identified as the cause of lipoid proteinosis. [2, 3, 6]
The exact mechanistic correlation between the genetic mutations described and the clinical manifestations of the disease remains unclear.
The Medscape Genomic Medicine Resource Center may be of interest.
Complications
Laryngeal involvement may lead to airway obstruction. Vocal cord involvement may lead to impaired speech.
Intracranial calcifications may result in seizures, behavioral changes, rage attacks, and dystonia.
The deposition of hyaline in the small bowel is reported to cause gastrointestinal bleeding.
Diagnostics
A pathognomonic finding on plain radiographs and CT scans of the brain is bilateral, intracranial, bean-shaped calcifications within the hippocampal region of the temporal lobes.
Porphyria should be excluded with appropriate blood and urine screening.
Skin biopsy of affected cutaneous or mucosal sites confirms the diagnosis in most cases.
Chatterjee et al report characteristic electroencephalogram findings in a 38-year-old male lipoid proteinosis patient. The findings include frontotemporal high amplitude slowing bilaterally. Notably, the patient was the product of a third-degree consanguineous union. [7]
Treatment
No cure is known. The disease follows a stable, chronic course but may fluctuate in intensity. No uniformly successful treatment is available.
Patient education
Patients should be educated about the risk of having affected offspring.
Pathophysiology
Lipoid proteinosis is a rare genodermatosis inherited in an autosomal recessive pattern. Clinical features are myriad, and the literature describes considerable variation in severity between affected patients. Heterozygous carriers have an apparently normal phenotype but may have subtle changes such as abnormal dentition.
In 2002, loss of function mutations in the gene encoding extracellular matrix protein 1 (ECM1) on band 1q21 were identified as the cause of lipoid proteinosis. [2, 8] Frameshift and nonsense mutations have been described throughout the gene, although exons 6 and 7 seem to be the most common locations. Mutations at these particular sites appear to have genotype-phenotype relevance. Patients with exon 7 mutations display slightly milder clinical features, while mutations in exon 6 result in a more severe phenotype.
The ECM1 gene product is a glycoprotein with functional roles in skin physiology and homeostasis. ECM1 is involved in keratinocyte differentiation in the epidermis and in regulation of basement membrane integrity, interstitial collagen fibril macroassembly, and growth factor binding in the dermis. The disease is characterized by deposits of hyalinelike material in skin, mucosa, and viscera and is also referred to as hyalinosis cutis et mucosae. The original designation, lipoid proteinosis, refers to the histologic features of the deposited material, which shows similarities to both lipid and protein, although no abnormalities in lipid metabolism have been identified.
ECM1 proteins are also expressed in a number of other tissues, including the placenta, heart, liver, small intestine, lungs, ovary, testes, prostate, pancreas, kidneys, skeletal muscle, and endothelial cells. Its role in wound healing, scarring, and aging is speculated but not yet defined.
The loss of normal function of ECM1 in lipoid proteinosis is associated with a wide range of clinical abnormalities due to infiltration of the skin and viscera with hyalinelike material. The histological correlate is diffuse dermal deposition of hyaline material, basement membrane thickening at the dermoepidermal junction and around adnexa and vessels, and epidermal hyperkeratosis. These findings suggest the strong influence of ECM1 on both epidermal and dermal physiology.
The eosinophilic hyaline material is deposited in all affected organs, although whether this is a primary or secondary phenomenon is unclear. The details of the genotype-phenotype correlation regarding the nature of the hyaline deposits are currently under investigation.
Epidemiology
More than 300 cases have been reported worldwide. [9] Incidence and prevalence figures are not available. Patients of European ancestry are most commonly affected, including South African descendants of German or Dutch immigrants. Large kindreds from South Africa have been traced back to a single German male who settled in South Africa in the 17th century.
No sex predilection is reported. Patients typically present in early childhood, but manifestations may be present at birth. Some cases may occur in adults. Adults may have subtle skin findings and may present with complications due to visceral deposition.
-
Characteristic beaded papules on the eyelid (moniliform blepharosis). Courtesy of Kenneth E. Greer, MD.
-
Waxy, yellow skin thickening and atrophic scarring. Courtesy of Kenneth E. Greer, MD.
-
Beaded papules on the upper labial mucosa. Courtesy of Kenneth E. Greer, MD.
-
Woody induration and depression of the tongue. Courtesy of Kenneth E. Greer, MD.
-
Waxy, infiltrated, yellowish skin with depressed, atrophic scarring. Courtesy of Kenneth E. Greer, MD.
-
Waxy skin with atrophic, depressed scars on the forehead. Courtesy of Kenneth E. Greer, MD.
-
Infiltrated, thickened skin with atrophic and hyperpigmented scarring in 2 brothers with lipoid proteinosis. Note the tongues, which are firm and woody, ulcerated, and unable to be completely protruded because of infiltration of the frenulum. Courtesy of Kenneth E. Greer, MD.
-
Adult male with lipoid proteinosis. His leonine facies appearance is a result of diffuse skin infiltration. Courtesy of Kenneth E. Greer, MD.


